- Research article
- Open access
- Published:
SARS-CoV-2 surveillance in Italy through phylogenomic inferences based on Hamming distances derived from pan-SNPs, -MNPs and -InDels
BMC Genomics volume 22, Article number: 782 (2021)
Abstract
Background
Faced with the ongoing global pandemic of coronavirus disease, the ‘National Reference Centre for Whole Genome Sequencing of microbial pathogens: database and bioinformatic analysis’ (GENPAT) formally established at the ‘Istituto Zooprofilattico Sperimentale dell’Abruzzo e del Molise’ (IZSAM) in Teramo (Italy) is in charge of the SARS-CoV-2 surveillance at the genomic scale. In a context of SARS-CoV-2 surveillance requiring correct and fast assessment of epidemiological clusters from substantial amount of samples, the present study proposes an analytical workflow for identifying accurately the PANGO lineages of SARS-CoV-2 samples and building of discriminant minimum spanning trees (MST) bypassing the usual time consuming phylogenomic inferences based on multiple sequence alignment (MSA) and substitution model.
Results
GENPAT constituted two collections of SARS-CoV-2 samples. The first collection consisted of SARS-CoV-2 positive swabs collected by IZSAM from the Abruzzo region (Italy), then sequenced by next generation sequencing (NGS) and analyzed in GENPAT (n = 1592), while the second collection included samples from several Italian provinces and retrieved from the reference Global Initiative on Sharing All Influenza Data (GISAID) (n = 17,201). The main results of the present work showed that (i) GENPAT and GISAID detected the same PANGO lineages, (ii) the PANGO lineages B.1.177 (i.e. historical in Italy) and B.1.1.7 (i.e. ‘UK variant’) are major concerns today in several Italian provinces, and the new MST-based method (iii) clusters most of the PANGO lineages together, (iv) with a higher dicriminatory power than PANGO lineages, (v) and faster that the usual phylogenomic methods based on MSA and substitution model.
Conclusions
The genome sequencing efforts of Italian provinces, combined with a structured national system of NGS data management, provided support for surveillance SARS-CoV-2 in Italy. We propose to build phylogenomic trees of SARS-CoV-2 variants through an accurate, discriminant and fast MST-based method avoiding the typical time consuming steps related to MSA and substitution model-based phylogenomic inference.
Introduction
The coronavirus disease 19 (COVID-19) responsible to the current pandemic is due to a novel coronavirus (CoV) named SARS-CoV-2 [1]. COVID-19 was firstly reported in humans during December 2019 in the city of Wuhan (Hubei Province, China) and the role of the Huanan seafood wholesale market in SARS-CoV-2 emergence is still uncertain [2,3,4]. At the date of the present study (May 2021), 222 countries were affected by the SARS-CoV-2 with 153,527,666 coronavirus cases, as well as 3,217,267 deaths, 680,364 daily new cases, and 9981 daily deaths [5]. With more than 1000 cases confirmed till the 1st March 2020, Italy was one of the first European countries to face the SARS-CoV-2 burden [6]. At the national level, the Italian Civil Protection Department counted today 4,044,762 total cases, 121,177 deaths, 3,492,679 recovered people and 430,906 active cases in Italy [7]. COVID-19 is mainly a respiratory infection, with the most common symptoms comprising fever, dry cough, and shortness of breath [8]. About 20% of infected patients may develop severe disease, and a small percentage (5%) may become critically ill [8]. Patients with severe COVID-19 disease may develop pneumonia or acute respiratory distress syndrome (ARDS), which is often fatal [9] and requires mechanical ventilation and treatment from intensive care unit [8].
CoVs harbor enveloped single-stranded RNA genomes with high plasticity [10] induced by a high-frequency RNA recombination [2, 3]. New genotypes have emerged from homologous RNA recombination, and novel genes have been acquired through heterogeneous RNA recombination with non-coronaviral donor RNAs [11]. SARS-CoV-2 is paradigmatic of these evolutionary mechanisms as there is compelling evidence that it emerged through recombination of SARS-related coronaviruses (SARSr-CoVs) as it was suggested for SARS-CoV-1 [2, 3, 12, 13]. Besides recombination events, the point mutations from replication errors drive also the SARS-CoV-2 evolution (i.e. single nucleotide polymorphisms (SNPs), multi-nucleotide polymorphisms (MNPs) and small insertions/deletions (InDels)) [14]. The likely SNP-based mutation rate of the SARS-CoV-2 (~ 10− 6 nt− 1 cycle− 1) is low compared to influenza virus (~ 3 × 10− 5 nt− 1 cycle− 1) or other RNA viruses [15]. In fact, the SARS-CoV-2 is able to repair part of duplication errors induced by the RNA-dependent RNA polymerases (RdRp) [16]. However, a SARS-CoV-2 population in one milliliter of sputum (i.e. around 107 RNAs/ml) with this likely mutation rate (~ 10− 6 nt− 1 cycle− 1) would harbor more than one mutation in every nucleotide [17], not mentioning that spreading over millions of individuals induces fast accumulation of mutations.
In addition to negative impacts of the SARS-CoV-2 on hospital workload [18], medical clinic organization [19], long-term health [20], small business [21], socio-economic system [22] and employment [23], the national health care systems have to face the need for thousands of laboratory tests per day [24]. The Veterinary Public Health Institutes, namely Istituti Zooprofilattici Sperimentali (IZS), perform the diagnosis of SARS-CoV-2 through testing nasopharyngeal swabs by RT-PCR on behalf of the Italian Ministry of Health [24]. In the face of the current COVID-19 crisis, the “National Reference Centre for Whole Genome Sequencing of microbial pathogens: database and bioinformatic analysis” (GENPAT) formally established at the IZS dell’Abruzzo e del Molise (IZSAM) in Teramo (G.U.R.I. 196, August 23, 2017), dedicates its developments to improve analytical workflows of SARS-CoV-2 sequences from routine surveillance activities.
Different international teams proposed analytical workflows to reconstruct SARS-CoV-2 genomes based on de novo assemblies [25, 26] and/or consensus sequences [27] from variant calling analysis [28,29,30] performed through mapping of reads [26, 28,29,30,31] against the reference genome Wuhan-Hu-1/2019. The resulted de novo assemblies and consensus sequences are commonly uploaded at the international level into the Global Initiative on Sharing All Influenza Data (GISAID) [32]. From the de novo assemblies or consensus sequences, the dedicated PANGOLIN tool performs the identification of SARS-CoV-2 lineages, so-called PANGO lineages [33], and has been adopted by the reference GISAID [32] because it allows sharing between laboratories of a common dynamic nomenclature of mutations associated with important functional evolution events [34]. Otherwise, these de novo assemblies and consensus sequences are usually aligned between each other through multiple sequence alignment (MSA) [28, 35,36,37] in order to perform substitution model-based phylogenomic inferences through maximum likelihood (ML) [28, 35] or Bayesian models [37]. The aligned de novo assemblies and consensus sequences can also be derived into variant calling format (i.e. VCF) [37]. Because the biological effects of variants (i.e. SNPs, MNPs and InDels, so-called genotypes in VCF files) are required for identifying mutations associated with important functional evolution events [34] and accordingly designing of SARS-CoV-2 vaccines [38], these VCF files or aligned sequences are typically input data of functional annotation of variants [28, 29, 37]. Even though de novo assembly [25, 39], mapping of reads [40,41,42,43] and variant calling analysis [44,45,46] are relatively fast processes, these SARS-CoV-2 workflows are currently limited by the time consuming steps aiming at performing MSA [47,48,49,50,51,52], then substitution model-based phylogenomic inferences [53,54,55]. In fact, the phylogenetic inferences based on MSA and substitution model can take many days or weeks depending of the available computing power, particularly when the dataset of samples includes several hundreds of genomes.
In the area of surveillance dedicated to bacteria including the genera Enterococcus [56], Mycoplasma [57], Pseudomonas [58], Mycobacterium [59], Brucella [60] and many others, coregenome and whole genome multi-locus sequence typing (cg/wgMLST) and corresponding schemes of alleles have been proposed to identify epidemiological relationships based on screening of alleles through several hundred or thousands of homologous genes, so-called loci [61]. In comparison with the so-called allele scheme, the combination of these MLST allele numbers from a single strain allows assignation of a MLST sequence type (ST) already shared between laboratories or a new one by default [62]. The output of cg/wgMLST methods from different analytical workflows (e.g. chewBBACA [63], SeqSphere+ [64], MLSTar [65], BIGSdb-Pasteur [66], Bionumerics [67]) are frequently used as input for a recent minimum spanning tree (MST) algorithm (“MSTree V2”) implemented in the workflow GrapeTree in order to visualize coregenome relationships among hundreds of thousands bacterial genomes [68]. Compared to the good practices aiming at building a phylogenomic tree based on MSA, a substitution model (i.e. JC69, K80, K81, F81, HKY85, T92, TN93, or GTR) and an inference approch (i.e. ML or Bayesian models) [69, 70], the construction of a MST with “MSTree V2” is theoretically faster because it implements a directional measure based on normalized asymmetric Hamming-like distances between pairs of STs assuming that one of the pair of STs is the ancestor of the other [68].
Considering that the SARS-CoV-2 surveillance needs an accurate, discriminant and fast assessment of epidemiological clusters from substantial amount of samples, the present study provides a variant calling analysis-based workflow, so-called GENPAT workflow, to accurately identify the PANGO lineages of SARS-CoV-2 samples in Italy and rapidly build highly discriminant MST bypassing the usual time consuming phylogenomic inferences based on multiple sequence alignment (MSA) and substitution model. More precisely, the present manuscript aims at answering the following questions:
-
Question i: Is the GENPAT workflow able to identify PANGO lineages compared to the reference GISAID?
-
Question ii: What do the sequencing effort in Italy and GENPAT workflow development in the Abruzzo region reveal about the PANGO lineages mainly circulating in Italian provinces?
-
Question iii: Does the MST-based clustering match the reference PANGO lineages and/or Italian provinces?
-
Question iv: What are the differences of discrimination power between the developed MST-based method and PANGO lineages?
-
Question v: What are the differences of speed between the developed MST-based method and the usual phylogenomic inferences based on MSA and substitution model?
Results
The questions described above (i.e. questions i, ii, iii, iv and v) were assessed with the GENPAT workflow combining the identification of PANGO lineages based on variant calling analysis (Fig. 1) and MST-based phylogenomic inference (Figs. 1 and 2), as well as two collections of SARS-CoV-2 samples isolated until April 2021 in Italy from GENPAT (Additional file 1) and GISAID (Additional file 2).
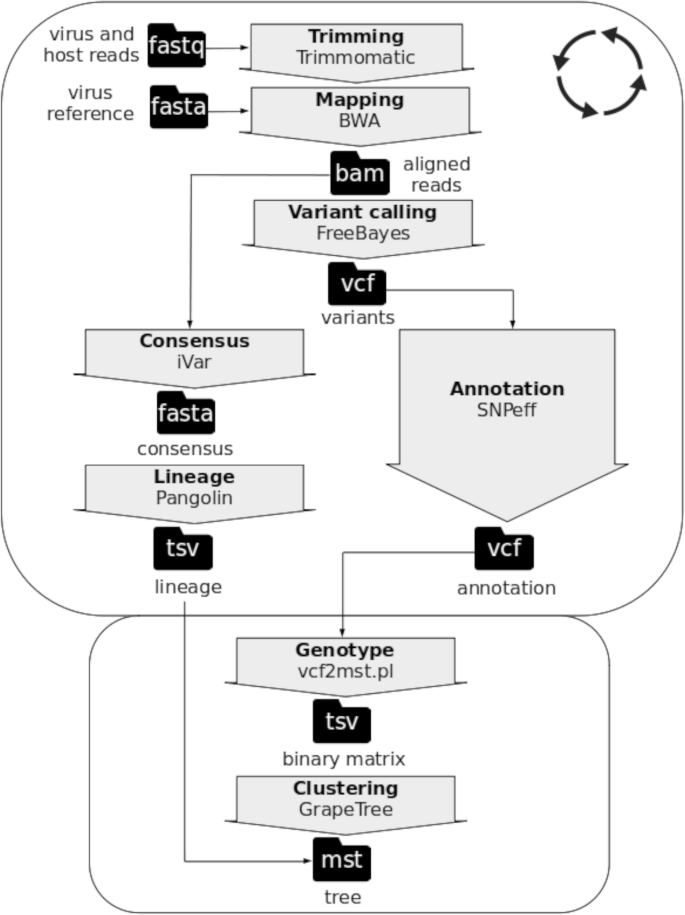
Sample dependent (rounded rectangle with circle of arrows) and dataset dependent (rounded rectangle without circle of arrows) steps of the workflow implemented in GENPAT to identify lineages of SARS-CoV-2 and build phylogenomic inference based on shotgun metagenomics paired-end read sequencing
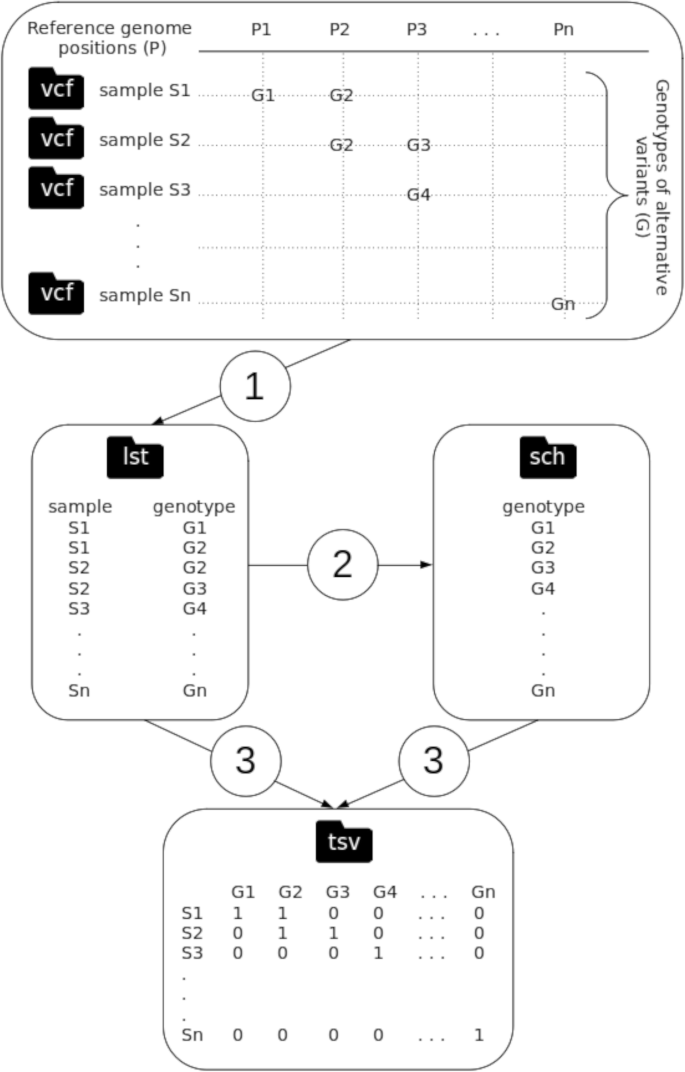
Algorithmic steps of the program “vcf2mst.pl” aiming to (1) derive functional annotations of variants (i.e. genotypes of pan-SNPs, -MNPs and -InDels) encoded in variant calling format (vcf) into lists of samples and genotypes (lst), (2) build a scheme of genotypes (sch) and (3) create a binary matrix of genotypes according to samples involved in reference genome based-variant calling analysis (tsv) for downstream inference of minimum spanning tree (MST)
GENPAT workflow ability to identify PANGO lineages in comparison to the reference GISAID
The GENPAT collection corresponds to samples isolated by IZSAM in the Abruzzo region, then sequenced by NGS and analyzed in GENPAT, while the GISAID collection corresponds to samples isolated in Italy and analyses by the reference GISAID. Comparing the collections GENPAT (Additional file 1, n = 1592 samples) and GISAID (Additional file 2, n = 17,201 samples), 1550 common SARS-CoV-2 samples presented identical PANGO lineages. In response to question i, the GENPAT workflow (Fig. 1) is as precise as the reference GISAID to identify PANGO lineages (Additional files 1 and 2).
Main PANGO lineages circulating in Italian provinces revealed by Italian genome sequencing activities and GENPAT workflow development
In view of the GISAID collection (Additional file 2, n = 17,201), the PANGO lineages B.1.1.7 (39%) and B.1.177 (17%) were the mostly identified in Italy (Fig. 3A), especially the province Napoli (24 and 12%) (n = 4184 and n = 2073) and, to a lesser extent, in the provinces of Venezia, Chieti, Bari, Trento and Teramo (Fig. 3B). With respect to the GISAID collection (Additional file 2), the lineages B.1.1.7 (39%) and B.1.177 (17%) were also the mostly detected in the Abruzzo region (Fig. 3C) and provinces of the Abruzzo region (Fig. 3D). Indeed, the PANGO lineages B.1.1.7 (62%) and B.1.177 (19%) were the mostly identified in the provinces of the Abruzzo region, namely Chieti (32 and 5%), L’Aquila (11 and 5%), Pescara (2% and 4‰) and Teramo (16 and 8%) (Table 1), among the SARS-CoV-2 samples from the GENPAT collection (Additional file 1, n = 1592). While the Napoli province produced the highest number of SARS-CoV-2 strain characterization in Italy (Fig. 3B, n = 10,372: 67%), the Chieti province presented the highest number of SARS-CoV-2 strains with an identified lineage in the Abruzzo region (Fig. 3B, n = 710: 45%). In response to question ii, the genome sequencing effort in Italy (i.e. GISAID collection) and GENPAT workflow developed in the Abruzzo region (i.e. GENPAT collection) revealed that PANGO lineages B.1.1.7 and B.1.177 have mainly circulating in Italy at the date of the present study.

Distributions of the 30 most frequent SARS-CoV-2 PANGO lineages in Italy centralized in GISAID and represented at the national level (A, n = 16,529); among the 6 most frequent provinces in Italy including Napoli (n = 10,115), Venezia (n = 1214), Chieti (n = 705), Bari (n = 661), Trento (n = 632) and Teroma (n = 501) (B, n = 13,828); among samples from the Abruzzo region mainly shotgun sequenced and analyzed in GENPAT (C, n = 1580); and inside the provinces of the Abruzzo region including Chieti (n = 705), Teromo (n = 501), l’Aquila (n = 320) and Percara (n = 54) (D)
Clustering by the MST-based method in comparison with the reference PANGO lineages
The GENPAT workflow provided MST-based clustering (Figs. 1 and 2) as exemplified by trees representing SARS-CoV-2 samples from the collections GENPAT (Fig. 4A and B, n = 1553) or GISAID (Fig. 4C and D, n = 15,451). For both collections GENPAT or GISAID including 30 and 176 PANGO lineages (Additional files 1 and 2), almost all the MST-based clusters matched with the reference PANGO lineages (Fig. 4A and C), but did not correspond specifically to Italian provinces (Fig. 4B and D). In reply to the question iii, the MST-based clustering implemented in the GENPAT workflow matched well the reference PANGO lineages without specific segregation of Italian provinces.

Pangenomic minimum spanning tree (MST) produced by the GENPAT workflow from SARS-CoV-2 samples isolated in provinces of the Abruzzo region and sequenced by GENPAT (A and B, n = 1553) or SARS-CoV-2 samples isolated in several Italian provinces and retrieved from GISAID (C and D, n = 15,451) with regard to lineages identified through PANGO dynamic nomenclature (A and C) or Italian provinces (B and D). The effective sample sizes are shown in square brackets
Discrimination power of the MST-based method in comparison with the reference PANGO lineages
The PANGO lineages B.1.1.7 and B.1.177 mostly identified in the Abruzzo province (Fig. 4A: 63 and 19%) and Italy (Fig. 4C: 40 and 18%), were both represented by multiple MST-based clusters (Fig. 3A and C). These multiple MST-based clusters were also observed for other less common PANGO lineages, such like B.1.1, B.1.177.8, B.1.1.420, B.1, P.1 and B.1.160 (Fig. 3A and C). In response to the question iii, the discrimination power of the MST-based method is higher than the reference PANGO lineages.
Speed of the MST-based method in comparison with the usual phylogenomic inferences based on MSA and substitution model
While MSA and substitution model-based phylogenomic inference would require several days to several weeks to reconstruct evolution history of several hundreds of genomes with a usual computing facility (i.e. server harboring 32 Go RAM and 32 core CPUs), GENPAT estimates that 30 s and 4 s were necessary to treat 1000 samples with the algorithms “vcf2mst.pl” and “MSTree V2”, respectively (Additional files 4 and 5). Concerning the question v, the MST-based method developed by GENPAT appears faster than the usual phylogenomic inferences based on MSA and substitution model.
Discussions
The correct detection of lineages (i) of concern (ii), as well as the accurate (iii), discriminant (iv) and fast (v) MST-based inference, are all in line with the SARS-CoV-2 surveillance requirements.
Accurate GENPAT identification of PANGO lineages
Due to exact match between PANGO lineages identified by GENPAT and the reference GISAID (Additional files 1 and 2, n = 1550), we recommend to identify SARS-CoV-2 lineages based on trimming, mapping, consensus building and lineage identification implemented in Trimmomatic [71], BWA [42], iVar [27] and PANGOLIN 2.0 [34], respectively (Fig. 1). Faced to the diversity of methods to identify SARS-CoV-2 variants based on variant calling analysis (i.e. SAMtools [28], Freebayes [29], GATK4 [30]) from mapping outcomes (i.e. Minimap [28], Minimap2 [29], BWA [30, 31], Bowtie2 [26]), we also encourage to pursue comparisons of these methods because the SARS-CoV-2 lineages are identified based on SNPs and InDels [34], while InDels are known to be more difficult to identify than SNPs [72].
Most frequent PANGO lineages B.1.177 and B.1.1.7 in Italy
Numerous lineages of SARS-CoV-2 emerged since the beginning of pandemic and, at the time of the manuscript preparation, three of them are today considered as global variants of concern: B.1.1.7, B.1.351, and P.1 (i.e. B.1.1.248 renamed B.1.1.28.1) [34]. These PANGO lineages B.1.1.7, B.1.351 and P1 were first detected in United Kingdom (UK) [73], South Africa [74] and Brazil [75], respectively. These lineages B.1.1.7, B.1.351, and P.1 replaced previous circulating variants in their original countries and spread to other countries in Europe, the Americas, and Asia [76]. There is an inordinate amount of concern for these three lineages because of likely reinfections due to reduced cross-protective immunity [77,78,79] and potential involvement in vaccine efficacy [80, 81]. While the PANGO lineage B.1.177 identified frequently in Abruzzo (Table 1) and Italy (Additional file 3) corresponds to one of the main lineage identified at the beginning of the pandemic event in Italy [82], the other frequently isolated PANGO lineage B.1.1.7 corresponds to the variant of concern called “UK variant” [75, 83,84,85,86]. In addition, GENPAT did not identified many samples corresponding to variants of concern named “South Africa” (B.1.351) [75, 83, 87, 88], “Japan-Brazil” (B.1.1.248 reclassified to B.1.1.28.1 - alias P.1) [75, 83, 87,88,89,90], “Nigeria” (B.1.1.207) [75, 91], “Denmark” (Y453F, 69–70deltaHV) [92, 93], “UK-Nigeria” (B.1.525) [94], and “Indian” (B.1.617) [95], neither in the Abruzzo region (Fig. 3C, Fig. 4AB, Table 1 and Additional file 1), or Italy (Fig. 3AB, Fig. 4CD, Additional files 2 and 3).
Clustering of the MST-based method in agreement with the PANGO lineages
To our knowledge, it is the first time that variant calling analysis [44,45,46] and MST-based method [68] are combined to infer phylogenomic history of SARS-CoV-2 samples. The adaptation of the MST-based method usually used after cg/wgMLST characterization of bacterial draft assemblies [56,57,58,59,60], to variant calling analysis widely used for SARS-CoV-2 investigation [28,29,30], allowed building of an efficient clustering workflow (Figs. 1 and 2), in almost complete agreement with the outcomes of the reference PANGO lineages [34]. In the foreseeable future, we would like to tag the MST clusters to provide a unified method to type SARS-CoV-2 lineages and build a phylogenomic inference at the same time.
High discriminatory power of the MST-based method
The fact that the two main PANGO lineages in Italy (i.e. B.1.1.7 and B.1.177) are constituted of multiple MST clusters, emphases that the proposed method (Figs. 1 and 2) has a higher discriminatory power than PANGO for SARS-CoV-2 typing (Fig. 3B and D). Indeed, the proposed MST-based phylogenomic inference is able to manage together pan-SNPs, -MNPs and -InDels (i.e. core and accessory variants) with respect to the reference genome. The present MST-based method (Figs. 1 and 2) is also able to build MST only based on genotypes from functional annotations of variants identified in specific SARS-CoV-2 epitopes. This useful option of the algorithm “vcf2mst.pl” aims at providing graphical warnings related to SARS-CoV-2 mutations acquired in regions known to be involved in immune responses [96]. Another useful feature of the algorithm “vcf2mst.pl” is a capacity to manage nucleotide (i.e. GENPAT collection) or amino acid (i.e. GISAID collection) patterns for users who only have one type of data.
Fast minimum spanning tree from function annotations of variants
In comparison to the time consuming steps (i.e. several days or weeks for thousands samples) aiming at performing MSA from de novo assemblies or consensus sequences [28, 31, 35, 36], as well as phylogenomic inferences based on substitution models [28, 35, 37], the Hamming distance-based method [68] developed in the present manuscript (Figs. 1 and 2) is very fast (i.e. tens of seconds to process thousands of samples). Even if this MST-based method does not root trees and does not take in account differences of evolution rates between lineages [68], this last allows fast graphical representation of SARS-CoV-2 spreading for surveillance requiring fast assessment of epidemiological clusters from substantial amount of samples (Fig. 4). In agreement with the World Health Organization (WHO), the rapid generation and sharing of virus genomic sequences will contribute to the understanding of transmission and the design of mitigation strategies [97]. Collaboration between public health bodies, data generators and analysts is essential to generate and use appropriately data for maximum public health benefit [97]. Concerning the research studies supporting that the SARS-CoV-2 emerged firstly from China in late 2019 [98,99,100,101] (i.e. firstly reported in December 2019 [4, 102, 103] with a plausible emergence between early October [104], or mid-October, and mid-November 2019 [105]), or from other countries at a similar period [106], or even earlier [107], the phylogenomic inference at a pangenomic scale based on MSA and substitution models [28, 35,36,37] remains the gold standard to confirm the geographical origin(s) of SARS-CoV-2 spreading, because our MST-based method does not root trees and does not integrate differences of evolution rates between lineages [68].
Conclusion
The main results of the present developments showed that (i) GENPAT and GISAID detected the same PANGO lineages, (ii) the PANGO lineages B.1.177 (i.e. historical in Italy) and B.1.1.7 (i.e. “UK variant”) are major concerns today in several Italian provinces, and the new MST-based method (iii) clusters most of the PANGO lineages together, (iv) with a higher discriminatory power than the PANGO lineages, (v) and faster than the usual phylogenomic methods based on MSA and substitution model. The genome sequencing efforts of Italian provinces, combined to a structured national management of metagenomics data, provided an accurate and fast answer supporting the system of SARS-CoV-2 surveillance in Italy. In addition, the outcomes of the present consortium involved in SARS-CoV-2 surveillance in Italy emphasized that the data sharing through GISAID is of paramount importance for supporting the international SARS-CoV-2 tracking.
Material and methods
A workflow was implemented in GENPAT during 2021 to identify SARS-CoV-2 lineages and build accurate, discriminant and fast phylogenomic inferences from several thousands of samples isolated in Italy based on shotgun metagenomics paired-end read sequencing (Fig. 1). In the present study, the adjective ‘discriminant’ refers to a high discriminatory power and the term ‘discriminatory power’ is defined as the ability of a molecular typing method to distinguish between two or more groups being assessed [108].
Collections of SARS-CoV-2 samples
Two collections of SARS-CoV-2 samples were established (i.e. metadata, lineages and functional annotation of variants). The first collection includes 1592 SARS-CoV-2 positive swab samples detected by IZSAM until April 2021 in the Abruzzo region (Italy), then sequenced by NGS and analyzed in GENPAT. Sequences were then systematically submitted by GENPAT to GISAID (https://www.gisaid.org/) [32]. The second collection harbors 17,201 samples isolated from different Italian regions and downloaded by GENPAT from GISAID in April 2021 [32]. While samples from the first collection were treated through the whole GENPAT workflow, those from the second collection were treated through the dataset dependent part of this workflow based on information retrieved from GISAID (Fig. 1).
SARS-CoV-2 detection and genome sequencing
Concerning the samples from the first collection, acquisition of sequencing data implied successively sampling (oropharyngeal swab transport medium or bronchoalveolar lavage), virus inactivation (PrimeStore® MTM, in BSL3 biocontainment laboratory), nucleic acid purification (MagMaxTM CORE from Thermofisher), real-time RT-PCR-based SARS-CoV-2 RNA detection (TaqManTM 2019-nCoV Assay Kit v1 or v2 from Thermofisher) [24], RNA reverse transcription through multiplexing PCR (primer scheme nCoV-2019/V1) following the ARTIC protocol (https://artic.network/) [109], cDNA purification (AMPure XP beads, Agencourt), cDNA quantification (Qubit dsDNA HS Assay Kit and Qubit fluorometer 2.0 from Thermofisher or QuantiFluor ONE dsDNA System from Promega and FLUOstar OMEGA from BMG Labtech), NGS library preparation (Illumina DNA Prep kit) and sequencing (2 × 150 bp: MiniSeq or NextSeq500 from Illumina).
Variant calling analysis
With the objective to avoid the time consuming MSA [47,48,49,50,51,52] and propose an accurate, discriminant and fast phylogenomic inference, the reference genome mapping [40,41,42,43], variant calling analysis [44,45,46] and functional annotation of variants (pan-SNPs, -MNPs and -InDels) [110, 111] were preferred to de novo assembly [25, 39] or consensus sequences [27]. More precisely, we implemented a mapping-based variant calling analysis including functional variant annotations based on Trimmomatic (version 0.36, parameters: illuminaclip:2:30:10, leading:25 trailing:25 slidingwindows:20:25, minlen: 36) [71], BWA (version 0.7.17, algorithm mem, default parameters) [42], FreeBayes (version 1.3.2, default parameters) [45] and SNPeff (version 4.3, default parameters) [110] implemented in Snippy (version 4.5.1, default parameters) [112] because this workflow is fast and already well packaged in Docker (Fig. 1). The usual SARS-CoV-2 reference genome Wuhan-Hu-1 (i.e. NC_045512) was used for read mapping and variant calling analysis.
Identification of lineages
Faced to the rareness of other tools dedicated to lineage identification of SARS-CoV-2 (Nextstrain [113], GISAID [114] and PhenoGraph-based [115]), the workflow PANGOLIN has been implemented in the GENPAT workflow (Fig. 1) to assign PANGO lineages with a multinomial logistic regression-based machine learning coupled to a dynamic nomenclature of mutations associated with important functional evolution events [34]. More in details, consensus sequences were derived from BWA-based read mapping [42] with the program iVar (version 1.3, parameters: minimum length of read to retain after trimming m = 1, minimum quality threshold for sliding window to pass q = 20) [27] before to be used as input of the workflow PANGOLIN (version 3.1.11, algorithm pangoLEARN, default parameters) [34] (Fig. 1). In brief, this PANGO dynamic nomenclature proposes to label major lineages with a letter starting from the earliest lineage A SARS-CoV-2 viruses closely related to the most recent common ancestor (MRCA) Wuhan/WH04/2020 (EPI_ISL_406801) isolated from the Hubei province (China) on 5 January 2020. The early representative SARS-CoV-2 sample of the lineage B was isolated on 26 December 2019: Wuhan-Hu-1 (GenBank accession no. MN908947). Then, the dynamic nomenclature assigns a numerical value for each descending lineage from either lineage A or B (e.g A.1 or B.2) following roles with corresponding criteria [34].
Phylogenomic inferences
Keeping in mind the objective to build phylogenomic trees matching the PANGO lineages, with high discriminatory power, and as fast as possible, we replaced the slow substitution model-based phylogenomic inference [53,54,55] by MST inferred with the algorithm “MSTree V2” implemented in GrapeTree (version 2.2, default parameters) [68] (Fig. 1). Similarly to cg/wgMLST workflows which use alleles of homologous genes to build MST, we propose in the present manuscript an algorithm called “vcf2mst.pl” to infer MST from functional annotation of variants (Fig. 1). This algorithm “vcf2mst.pl” (version 1.0, default parameters) uses sample dependent VCF files from upstream reference genome based-variant calling analysis (Fig. 1) to build a binary matrix of genotypes representing unique functional annotations of variants encoded in these VCF files (Fig. 2). The three main steps of this algorithm “vcf2mst.pl” aims to (1) derive functional annotations of variants (i.e. genotypes) encoded in variant calling format (vcf) into lists of samples and genotypes (lst), (2) build a scheme of genotypes (sch) and (3) create a binary matrix of genotypes according to samples of interest (Fig. 2). This algorithm “vcf2mst.pl” encodes the unique genotypes of SNPs and MNPs according to the nucleotide pattern “reference genotype - position - alternative genotype” (e.g. snp: C241T), while the unique genotypes of InDels are encoded following the nucleotide pattern “position - reference genotype - alternative genotype” (e.g. ins:11287-G-GTCTGGTTTT or del:11287-GTCTGGTTTT-G). In contrast, the unique genotypes from GISAID (i.e. ZAPPO_GISAID_VCF) are encodes following the amino acid patterns “gene name _ reference amino acid _ position _ alternative amino acid” for SNPs and MNPs (e.g. NSP12_P323L or Spike_D614G), “gene name _ ins _ position _ amino acid” for insertions (e.g. NSP6_ins35VL) and “gene name _ amino acid _ position _ del” for deletions (e.g. NSP1_M85del). The proposed MST-based phylogenomic inference is able to manage together pan-SNPs, -MNPs and -InDels (i.e. core and accessory variants) with respect to the reference genome, because the presence of alternative genotype is encodes “1”, while the absence of alternative genotype is encodes “0”.
Availability of data and materials
Metadata and consensus sequences of all the SARS-CoV-2 are available from GISAID (https://www.gisaid.org/) at the accession numbers described in supplementary information. The algorithm “vcf2mst.pl” is available in GitHub (https://github.com/genpat-it/vcf2mst).
Abbreviations
- ARDS:
-
Acute respiratory distress syndrome
- cg/wgMLST:
-
Coregenome and whole genome multi-locus sequence typing
- CoV:
-
Coronavirus
- COVID-19:
-
Coronavirus disease 19
- GATK4:
-
Genomic analysis toolkit
- GENPAT:
-
Whole Genome Sequencing of microbial pathogens: data-base and bioinformatics analysis
- GISAID:
-
Global Initiative on Sharing All Influenza Data
- IZSAM:
-
Istituto Zooprofilattico Sperimentale dell’Abruzzo e del Molise Giuseppe Caporale
- ML:
-
Maximum likelihood
- MSA:
-
Multiple sequence alignment
- MST:
-
Minimum spanning trees
- NGS:
-
Next generation sequencing
- NRC:
-
National Reference Centre
- RdRp:
-
RNA-dependent RNA polymerases
- SARSr-CoVs:
-
SARS-related coronaviruses
- SARS-rCoV:
-
Severe acute respiratory syndrome-related virus
- ST:
-
Sequence type
- UK:
-
United Kingdom
- VCF:
-
Variant calling format
- WHO:
-
World Health Organization
References
Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5(4):536–44. https://doi.org/10.1038/s41564-020-0695-z.
Decaro N, Lorusso A. Novel human coronavirus (SARS-CoV-2): a lesson from animal coronaviruses. Vet Microbiol. 2020;244:108693. https://doi.org/10.1016/j.vetmic.2020.108693.
Lorusso A, Calistri P, Petrini A, Savini G, Decaro N. Novel coronavirus (SARS-CoV-2) epidemic: a veterinary perspective. Vet Ital. 2020;56(1):5–10. https://doi.org/10.12834/VetIt.2173.11599.1.
Wu F, Zhao S, Yu B, Chen Y-M, Wang W, Song Z-G, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–9. https://doi.org/10.1038/s41586-020-2008-3.
WorldOmeter. Covid-19 coronavirus pandemic. 2021. https://www.worldometers.info/coronavirus/.
Di Giallonardo F, Duchene S, Puglia I, Curini V, Profeta F, Cammà C, et al. Genomic epidemiology of the first wave of SARS-CoV-2 in Italy. Viruses. 2020;12(12):1438. https://doi.org/10.3390/v12121438.
Mossotto F. Elaboration and data for Feb 19 to 23 (last update 03 May 2021). Powered HCL Workload Autom; 2021.
Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med. 2020;8(5):475–81. https://doi.org/10.1016/S2213-2600(20)30079-5.
Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute respiratory distress syndrome. Nat Rev Dis Primer. 2019;5(1):18. https://doi.org/10.1038/s41572-019-0069-0.
Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet Lond Engl. 2020;395(10224):565–74. https://doi.org/10.1016/S0140-6736(20)30251-8.
Luytjes W, Bredenbeek PJ, Noten AF, Horzinek MC, Spaan WJ. Sequence of mouse hepatitis virus A59 mRNA 2: indications for RNA recombination between coronaviruses and influenza C virus. Virology. 1988;166(2):415–22. https://doi.org/10.1016/0042-6822(88)90512-0.
Boni MF, Lemey P, Jiang X, Lam TT-Y, Perry BW, Castoe TA, et al. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat Microbiol. 2020;5(11):1408–17. https://doi.org/10.1038/s41564-020-0771-4.
Hu B, Zeng L-P, Yang X-L, Ge X-Y, Zhang W, Li B, et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017;13(11):e1006698. https://doi.org/10.1371/journal.ppat.1006698.
Kosuge M, Furusawa-Nishii E, Ito K, Saito Y, Ogasawara K. Point mutation bias in SARS-CoV-2 variants results in increased ability to stimulate inflammatory responses. Sci Rep. 2020;10(1):17766. https://doi.org/10.1038/s41598-020-74843-x.
Sanjuán R, Nebot MR, Chirico N, Mansky LM, Belshaw R. Viral mutation rates. J Virol. 2010;84(19):9733–48. https://doi.org/10.1128/JVI.00694-10.
Pachetti M, Marini B, Benedetti F, Giudici F, Mauro E, Storici P, et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J Transl Med. 2020;18(1):179. https://doi.org/10.1186/s12967-020-02344-6.
Bar-On YM, Flamholz A, Phillips R, Milo R. SARS-CoV-2 (COVID-19) by the numbers. eLife. 2020;9:e57309. https://doi.org/10.7554/eLife.57309.
Klein MG, Cheng CJ, Lii E, Mao K, Mesbahi H, Zhu T, et al. COVID-19 Models for Hospital Surge Capacity Planning: A Systematic Review. Disaster Med Public Health Prep. 2020;10:1–8.
Ather A, Patel B, Ruparel NB, Diogenes A, Hargreaves KM. Coronavirus disease 19 (COVID-19): implications for clinical dental care. J Endod. 2020;46(5):584–95. https://doi.org/10.1016/j.joen.2020.03.008.
Huang C, Huang L, Wang Y, Li X, Ren L, Gu X, et al. 6-month consequences of COVID-19 in patients discharged from hospital: a cohort study. Lancet. 2021;397(10270):220–32. https://doi.org/10.1016/S0140-6736(20)32656-8.
Bartik AW, Bertrand M, Cullen Z, Glaeser EL, Luca M, Stanton C. The impact of COVID-19 on small business outcomes and expectations. Proc Natl Acad Sci. 2020;117(30):17656–66. https://doi.org/10.1073/pnas.2006991117.
Nicola M, Alsafi Z, Sohrabi C, Kerwan A, Al-Jabir A, Iosifidis C, et al. The socio-economic implications of the coronavirus pandemic (COVID-19): a review. Int J Surg. 2020;78:185–93. https://doi.org/10.1016/j.ijsu.2020.04.018.
Fana M, Torrejón Pérez S, Fernández-Macías E. Employment impact of Covid-19 crisis: from short term effects to long terms prospects. J Ind Bus Econ. 2020;47(3):391–410. https://doi.org/10.1007/s40812-020-00168-5.
Lorusso A, Calistri P, Mercante MT, Monaco F, Portanti O, Marcacci M, et al. A “one-health” approach for diagnosis and molecular characterization of SARS-CoV-2 in Italy. One Health. 2020;10:100135. https://doi.org/10.1016/j.onehlt.2020.100135.
Meleshko D, Hajirasouliha I, Korobeynikov A. coronaSPAdes: from biosynthetic gene clusters to RNA viral assemblies. Preprint. Bioinformatics. 2020. https://doi.org/10.1101/2020.07.28.224584.
Hu T, Li J, Zhou H, Li C, Holmes EC, Shi W. Bioinformatics resources for SARS-CoV-2 discovery and surveillance. Brief Bioinform. 2021;22(2):631–41. https://doi.org/10.1093/bib/bbaa386.
Grubaugh ND, Gangavarapu K, Quick J, Matteson NL, De Jesus JG, Main BJ, et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019;20(1):8. https://doi.org/10.1186/s13059-018-1618-7.
Laamarti M, Alouane T, Kartti S, Chemao-Elfihri MW, Hakmi M, Essabbar A, et al. Large scale genomic analysis of 3067 SARS-CoV-2 genomes reveals a clonal geo-distribution and a rich genetic variations of hotspots mutations. PLoS One. 2020;15(11):e0240345. https://doi.org/10.1371/journal.pone.0240345.
Pfefferle S, Günther T, Kobbe R, Czech-Sioli M, Nörz D, Santer R, et al. SARS Coronavirus-2 variant tracing within the first Coronavirus Disease 19 clusters in northern Germany. Clin Microbiol Infect Off Publ Eur Soc Clin Microbiol Infect Dis. 2021;27:130.e5–8.
Hufsky F, Lamkiewicz K, Almeida A, Aouacheria A, Arighi C, Bateman A, et al. Computational strategies to combat COVID-19: useful tools to accelerate SARS-CoV-2 and coronavirus research. Brief Bioinform. 2021;22(2):642–63. https://doi.org/10.1093/bib/bbaa232.
Chen S, He C, Li Y, Li Z, Melançon CE. A computational toolset for rapid identification of SARS-CoV-2, other viruses and microorganisms from sequencing data. Brief Bioinform. 2021;22(2):924–35. https://doi.org/10.1093/bib/bbaa231.
Shu Y, McCauley J. GISAID: global initiative on sharing all influenza data - from vision to reality. Euro Surveill Bull Eur Sur Mal Transm Eur Commun Dis Bull. 2017;22(13). https://doi.org/10.2807/1560-7917.ES.2017.22.13.30494.
Rambaut A, Holmes EC, O’Toole Á, Hill V, McCrone JT, Ruis C, et al. Addendum: a dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol. 2021;6(3):415. https://doi.org/10.1038/s41564-021-00872-5.
Rambaut A, Holmes EC, O’Toole Á, Hill V, McCrone JT, Ruis C, et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol. 2020;5(11):1403–7. https://doi.org/10.1038/s41564-020-0770-5.
Adebalİ O, Bİrcan A, Çİrcİ D, İŞlek B, KilinÇ Z, SelÇuk B, et al. Phylogenetic analysis of SARS-CoV-2 genomes in Turkey. Turk J Biol Turk Biyol Derg. 2020;44:146–56.
Saha I, Ghosh N, Maity D, Sharma N, Mitra K. Inferring the genetic variability in Indian SARS-CoV-2 genomes using consensus of multiple sequence alignment techniques. Infect Genet Evol. 2020;85:104522. https://doi.org/10.1016/j.meegid.2020.104522.
Bindayna KM, Crinion S. Variant analysis of SARS-CoV-2 genomes in the Middle East. Microb Pathog. 2021;153:104741. https://doi.org/10.1016/j.micpath.2021.104741.
Jeon JS, Won YH, Kim IK, Ahn JH, Shin OS, Kim JH, et al. Analysis of single nucleotide polymorphism among varicella-zoster virus and identification of vaccine-specific sites. Virology. 2016;496:277–86. https://doi.org/10.1016/j.virol.2016.06.017.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–52. https://doi.org/10.1038/nbt.1883.
Li H. Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinforma Oxf Engl. 2016;32(14):2103–10. https://doi.org/10.1093/bioinformatics/btw152.
Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinforma Oxf Engl. 2018;34(18):3094–100. https://doi.org/10.1093/bioinformatics/bty191.
Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25(14):1754–60. https://doi.org/10.1093/bioinformatics/btp324.
Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9(4):357–9. https://doi.org/10.1038/nmeth.1923.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. https://doi.org/10.1093/bioinformatics/btp352.
Richter F, Morton SU, Qi H, Kitaygorodsky A, Wang J, Homsy J, et al. Whole Genome De Novo Variant Identification with FreeBayes and Neural Network Approaches. preprint. Genomics. 2020. https://doi.org/10.1101/2020.03.24.994160.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. https://doi.org/10.1101/gr.107524.110.
Huddleston J, Hadfield J, Sibley T, Lee J, Fay K, Ilcisin M, et al. Augur: a bioinformatics toolkit for phylogenetic analyses of human pathogens. J Open Source Softw. 2021;6(57):2906. https://doi.org/10.21105/joss.02906.
Katoh K. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30(14):3059–66. https://doi.org/10.1093/nar/gkf436.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7. https://doi.org/10.1093/nar/gkh340.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–80. https://doi.org/10.1093/nar/22.22.4673.
Sievers F, Higgins DG. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol Biol Clifton NJ. 2014;1079:105–16. https://doi.org/10.1007/978-1-62703-646-7_6.
Saha I, Ghosh N, Maity D, Sharma N, Sarkar JP, Mitra K. Genome-wide analysis of Indian SARS-CoV-2 genomes for the identification of genetic mutation and SNP. Infect Genet Evol J Mol Epidemiol Evol Genet Infect Dis. 2020;85:104457. https://doi.org/10.1016/j.meegid.2020.104457.
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020;37(5):1530–4. https://doi.org/10.1093/molbev/msaa015.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3. https://doi.org/10.1093/bioinformatics/btu033.
Remco B, Heled J, Kühnert D, Vaughan T, Wu CH, Xie D, et al. “BEAST 2: a software platform for Bayesian evolutionary analysis.” Edited by Andreas. Prlic PLoS Comput Biol. 2014;10(4):e1003537. https://doi.org/10.1371/journal.pcbi.1003537.
Neumann B, Prior K, Bender JK, Harmsen D, Klare I, Fuchs S, et al. A Core genome multilocus sequence typing scheme for Enterococcus faecalis. J Clin Microbiol. 2019;57(3). https://doi.org/10.1128/JCM.01686-18.
Ghanem M, Wang L, Zhang Y, Edwards S, Lu A, Ley D, et al. Core genome multilocus sequence typing: a standardized approach for molecular typing of Mycoplasma gallisepticum. J Clin Microbiol. 2018;56(1). https://doi.org/10.1128/JCM.01145-17.
de Sales RO, Migliorini LB, Puga R, Kocsis B, Severino P. A Core genome multilocus sequence typing scheme for Pseudomonas aeruginosa. Front Microbiol. 2020;11:1049. https://doi.org/10.3389/fmicb.2020.01049.
Jones RC, Harris LG, Morgan S, Ruddy MC, Perry M, Williams R, et al. Phylogenetic analysis of Mycobacterium tuberculosis strains in Wales by use of Core genome multilocus sequence typing to analyze whole-genome sequencing data. J Clin Microbiol. 2019;57(6). https://doi.org/10.1128/JCM.02025-18.
Sankarasubramanian J, Vishnu US, Gunasekaran P, Rajendhran J. Development and evaluation of a core genome multilocus sequence typing (cgMLST) scheme for Brucella spp. Infect Genet Evol J Mol Epidemiol Evol Genet Infect Dis. 2019;67:38–43. https://doi.org/10.1016/j.meegid.2018.10.021.
Soucy SM, Huang J, Gogarten JP. Horizontal gene transfer: building the web of life. Nat Rev Genet. 2015;16(8):472–82. https://doi.org/10.1038/nrg3962.
Tse CW, Curreem SO, Cheung I, Tang BS, Leung K-W, Lau SK, et al. A novel MLST sequence type discovered in the first fatal case of Laribacter hongkongensis bacteremia clusters with the sequence types of other human isolates. Emerg Microbes Infect. 2014;3(1):e41–7. https://doi.org/10.1038/emi.2014.39.
Silva M, Machado MP, Silva DN, Rossi M, Moran-Gilad J, Santos S, et al. chewBBACA: a complete suite for gene-by-gene schema creation and strain identification. Microb Genomics. 2018;4(3). https://doi.org/10.1099/mgen.0.000166.
Ruppitsch W, Pietzka A, Prior K, Bletz S, Fernandez HL, Allerberger F, et al. Defining and evaluating a Core genome multilocus sequence typing scheme for whole-genome sequence-based typing of listeria monocytogenes. J Clin Microbiol. 2015;53(9):2869–76. https://doi.org/10.1128/JCM.01193-15.
Ferrés I, Iraola G. MLSTar: automatic multilocus sequence typing of bacterial genomes in R. PeerJ. 2018;6:e5098. https://doi.org/10.7717/peerj.5098.
Ragon M, Wirth T, Hollandt F, Lavenir R, Lecuit M, Le Monnier A, et al. A new perspective on listeria monocytogenes evolution. PLoS Pathog. 2008;4(9):e1000146. https://doi.org/10.1371/journal.ppat.1000146.
Radomski N, Cadel-Six S, Cherchame E, Felten A, Barbet P, Palma F, et al. A simple and robust statistical method to define genetic relatedness of samples related to outbreaks at the genomic scale - application to retrospective Salmonella foodborne outbreak investigations. Front Microbiol. 2019;10:2413. https://doi.org/10.3389/fmicb.2019.02413.
Zhou Z, Alikhan N-F, Sergeant MJ, Luhmann N, Vaz C, Francisco AP, et al. GrapeTree: visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res. 2018;28(9):1395–404. https://doi.org/10.1101/gr.232397.117.
Yang Z, Rannala B. Molecular phylogenetics: principles and practice. Nat Rev Genet. 2012;13(5):303–14. https://doi.org/10.1038/nrg3186.
Kapli P, Yang Z, Telford MJ. Phylogenetic tree building in the genomic age. Nat Rev Genet. 2020;21(7):428–44. https://doi.org/10.1038/s41576-020-0233-0.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinforma Oxf Engl. 2014;30(15):2114–20. https://doi.org/10.1093/bioinformatics/btu170.
Albers CA, Lunter G, MacArthur DG, McVean G, Ouwehand WH, Durbin R. Dindel: accurate indel calls from short-read data. Genome Res. 2011;21(6):961–73. https://doi.org/10.1101/gr.112326.110.
Davies NG, Abbott S, Barnard RC, Jarvis CI, Kucharski AJ, Munday JD, et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science. 2021;372(6538):eabg3055.
Wagner E, Zaiser A, Leitner R, Quijada NM, Pracser N, Pietzka A, et al. Virulence characterization and comparative genomics of listeria monocytogenes sequence type 155 strains. BMC Genomics. 2020;21(1):847. https://doi.org/10.1186/s12864-020-07263-w.
CDC. Emerging SARS-CoV-2 Variants. Cent Dis Control Prev. Retrieved 16 March 2021. https://www.cdc.gov/coronavirus/2019-ncov/more/science-and-research/scientific-brief-emerging-variants.html.
O’Toole A, Hill V, Pybus OG, Watts A, Bogoch II, Khan K, et al. Tracking the international spread of SARS-CoV-2 lineages B.1.1.7 and B.1.351/501Y-V2. Available Online Accessed 1 March 2021. https://virological.org/t/tracking-the-international-spread-of-sars-cov-2-lineages-b-1-1-7-and-b-1-351-501y-v2/592.
Cristina Resende P, Felipe Bezerra J, Teixeira de Vasconcelos RH, Arantes I, Appolinario L, Carolina Mendonça A, et al. Spike E484K mutation in the first SARS-CoV-2 reinfection case confirmed in Brazil, 2020. Available Online Accessed 1 March 2021. https://virological.org/t/spike-e484k-mutation-in-the-first-sars-cov-2-reinfection-case-confirmed-in-brazil-2020/584.
Naveca F, da Costa C, Nascimento V, Souza V, Corado A, Nascimento F, et al. SARS-CoV-2 reinfection by the new Variant of Concern (VOC) P.1 in Amazonas, Brazil. Available Online Accessed 1 March 2021. https://virological.org/t/sars-cov-2-reinfection-by-the-new-variant-of-concern-voc-p-1-in-amazonas-brazil/596.
Wibmer CK, Ayres F, Hermanus T, Madzivhandila M, Kgagudi P, Oosthuysen B, et al. SARS-CoV-2 501Y.V2 escapes neutralization by south African COVID-19 donor plasma. Nat Med. 2021;27(4):622–5. https://doi.org/10.1038/s41591-021-01285-x.
Williams TC, Burgers WA. SARS-CoV-2 evolution and vaccines: cause for concern? Lancet Respir Med. 2021;9(4):333–5. https://doi.org/10.1016/S2213-2600(21)00075-8.
Xie X, Liu Y, Liu J, Zhang X, Zou J, Fontes-Garfias CR, et al. Neutralization of SARS-CoV-2 spike 69/70 deletion, E484K and N501Y variants by BNT162b2 vaccine-elicited sera. Nat Med. 2021;27(4):620–1. https://doi.org/10.1038/s41591-021-01270-4.
Di Giallonardo F, Puglia I, Curini V, Cammà C, Mangone I, Calistri P, et al. Emergence and Spread of SARS-CoV-2 Lineages B.1.1.7 and P.1 in Italy. Viruses. 2021;13:794.
ECDC (21 January 2021). Risk related to the spread of new SARS-CoV-2 variants of concern in the EU/EEA - first update. Eur Cent Dis Prev Control Retrieved 16 March 2021. https://www.ecdc.europa.eu/en/publications-data/covid-19-risk-assessment-spread-new-variants-concern-eueea-first-update.
Chand M, Hopkins S, Dabrera G, Achison C, Barclay W, Ferguson N, et al. Potential impact of spike variant N501Y. :6.
Challen R, Brooks-Pollock E, Read JM, Dyson L, Tsaneva-Atanasova K, Danon L. Risk of mortality in patients infected with SARS-CoV-2 variant of concern 202012/1: matched cohort study. BMJ. 2021;372:n579. https://doi.org/10.1136/bmj.n579.
Volz E, Mishra S, Chand M, Barrett JC, Johnson R, Geidelberg L, et al. Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. preprint. Infect Dis (except HIV/AIDS). 2021. https://doi.org/10.1101/2020.12.30.20249034.
Kupferschmidt K. New coronavirus variants could cause more reinfections, require updated vaccines. Science. 2021. https://doi.org/10.1126/science.abg6028.
Kupferschmidt K. New mutations raise specter of ‘immune escape. Science. 2021;371(6527):329–30. https://doi.org/10.1126/science.371.6527.329.
National Institute of Infectious Diseases (NIID), Japan. Brief report: New Variant Strain of SARS-CoV-2 Identified in Travelers from Brazil. Retrieved 16 March 2021. https://www.niid.go.jp/niid/en/2019-ncov-e/10108-covid19-33-en.html.
Voloch CM, da Silva Francisco R Jr, de Almeida LG, Cardoso CC, Brustolini OJ, Gerber AL, et al. Genomic characterization of a novel SARS-CoV-2 lineage from Rio de Janeiro, Brazil. preprint. Genet Genomic Med. 2020. https://doi.org/10.1101/2020.12.23.20248598.
Happi C, Ihekweazu C, Nkengasong J, Eniola Oluniyi P, Olawoye I. Detection of SARS-CoV-2 P681H Spike Protein Variant in Nigeria. Available Online Accessed 1 Dec 2020. https://virological.org/t/detection-of-sars-cov-2-p681h-spike-protein-variant-in-nigeria/567.
Koopmans M. SARS-CoV-2 and the human-animal interface: outbreaks on mink farms. Lancet Infect Dis. 2021;21(1):18–9. https://doi.org/10.1016/S1473-3099(20)30912-9.
ECDC. Detection of new SARS-CoV-2 variants related to mink. 2020. Retrieved 16 March 2021. https://www.ecdc.europa.eu/sites/default/files/documents/RRA-SARS-CoV-2-in-mink-12-nov-2020.pdf.
PHE. Variants: distribution of cases data updated 16 March 2021. 2021. Retrieved 16 March 2021 . https://www.gov.uk/government/publications/covid-19-variants-genomically-confirmed-case-numbers/variants-distribution-of-cases-data.
Yadav PD, Sapkal GN, Abraham P, Ella R, Deshpande G, Patil DY, et al. Neutralization of variant under investigation B.1.617 with sera of BBV152 vaccinees. Preprint. Immunology. 2021. https://doi.org/10.1101/2021.04.23.441101.
Shomuradova AS, Vagida MS, Sheetikov SA, Zornikova KV, Kiryukhin D, Titov A, et al. SARS-CoV-2 Epitopes Are Recognized by a Public and Diverse Repertoire of Human T Cell Receptors. Immunity. 2020;53:1245–1257.e5.
WHO. Genomic sequencing of SARS-CoV-2. A guide to implementation for maximum impact on public health. 8 January 2021. 2021;CC BY-NC-SA 3.0 IGO:1–80.
Worobey M, Pekar J, Larsen BB, Nelson MI, Hill V, Joy JB, et al. The emergence of SARS-CoV-2 in Europe and North America. Science. 2020;370(6516):564–70. https://doi.org/10.1126/science.abc8169.
Alteri C, Cento V, Piralla A, Costabile V, Tallarita M, Colagrossi L, et al. Genomic epidemiology of SARS-CoV-2 reveals multiple lineages and early spread of SARS-CoV-2 infections in Lombardy. Italy Nat Commun. 2021;12(1):434. https://doi.org/10.1038/s41467-020-20688-x.
Chinazzi M, Davis JT, Ajelli M, Gioannini C, Litvinova M, Merler S, et al. The effect of travel restrictions on the spread of the 2019 novel coronavirus (COVID-19) outbreak. Science. 2020;368(6489):395–400. https://doi.org/10.1126/science.aba9757.
Al-Salem W, Moraga P, Ghazi H, Madad S, Hotez PJ. The emergence and transmission of COVID-19 in European countries, 2019–2020: a comprehensive review of timelines, cases and containment. Int Health. 2021;13(5):383–98. https://doi.org/10.1093/inthealth/ihab037.
Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–3. https://doi.org/10.1038/s41586-020-2012-7.
Zhang Y-Z, Holmes EC. A genomic perspective on the origin and emergence of SARS-CoV-2. Cell. 2020;181(2):223–7. https://doi.org/10.1016/j.cell.2020.03.035.
Roberts DL, Rossman JS, Jarić I. Dating first cases of COVID-19. PLoS Pathog. 2021;17(6):e1009620. https://doi.org/10.1371/journal.ppat.1009620.
Pekar J, Worobey M, Moshiri N, Scheffler K, Wertheim JO. Timing the SARS-CoV-2 index case in Hubei Province. Preprint. Evol Biol. 2020. https://doi.org/10.1101/2020.11.20.392126.
La Rosa G, Mancini P, Bonanno Ferraro G, Veneri C, Iaconelli M, Bonadonna L, et al. SARS-CoV-2 has been circulating in northern Italy since December 2019: evidence from environmental monitoring. Sci Total Environ. 2021;750:141711. https://doi.org/10.1016/j.scitotenv.2020.141711.
Apolone G, Montomoli E, Manenti A, Boeri M, Sabia F, Hyseni I, et al. Unexpected detection of SARS-CoV-2 antibodies in the prepandemic period in Italy. Tumori J. 2020;107(5):446–51.
Blanc DS, Hauser PM, Francioli P, Bille J. Molecular typing methods and their discriminatory power. Clin Microbiol Infect. 1998;4(2):61–3. https://doi.org/10.1111/j.1469-0691.1998.tb00356.x.
Itokawa K, Sekizuka T, Hashino M, Tanaka R, Kuroda M. Disentangling primer interactions improves SARS-CoV-2 genome sequencing by multiplex tiling PCR. PLoS One. 2020;15(9):e0239403. https://doi.org/10.1371/journal.pone.0239403.
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w 1118 ; iso-2; iso-3. Fly (Austin) 2012;6:80–92. https://doi.org/10.4161/fly.19695.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. https://doi.org/10.1093/nar/gkq603.
Palma F, Brauge T, Radomski N, Mallet L, Felten A, Mistou M-Y, et al. Dynamics of mobile genetic elements of listeria monocytogenes persisting in ready-to-eat seafood processing plants in France. BMC Genomics. 2020;21(1):130. https://doi.org/10.1186/s12864-020-6544-x.
Bedford T, Hodcroft EB, Neher RA. Updated Nextstrain SARS-CoV-2 clade naming strategy. Retrieved 16 March 2021. https://nextstrain.org/blog/2021-01-06-updated-SARS-CoV-2-clade-naming.
GISAID. Clade and lineage nomenclature aids in genomic epidemiology studies of active hCoV-19 viruses. 2021. https://www.gisaid.org/references/statements-clarifications/clade-and-lineage-nomenclature-aids-in-genomic-epidemiology-of-active-hcov-19-viruses/.
Yang Z-K, Pan L, Zhang Y, Luo H, Gao F. Data-driven identification of SARS-CoV-2 subpopulations using PhenoGraph and binary-coded genomic data. Brief Bioinform. 2021;00(00):1–11.
Acknowledgements
We thank especially the Italian Ministry of Health for supporting in the acquisition of high-performance computing resources.
Funding
The study was funded by the European Union’s Horizon 2020 Research and Innovation program under grant agreement No 773830: One Health European Joint Program and by the Italian Ministry of Health IZSAM 05/20 Ricerca Corrente 2020 “PanCO: epidemiologia e patogenesi dei coronavirus umani ed animali”. The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript. Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the IZSAM.
Author information
Authors and Affiliations
Contributions
PC, AL and CC implemented the wet-lab procedure of sequencing, while ADP, NR and IM implemented the dry-lab procedure of NGS data analysis. All authors from the wet- (PC, AL and CC) and dry-lab (ADP, NR and IM) have made substantial contributions to the conception and design of the work, as well as to the interpretation of data. PC, AL and CC were involved in the acquisition of samples, metadata and NGS data. IM integrated the variant calling-based workflow in the GENPAT system with Python. ADP developed the Pearl-based algorithm “vcf2mst.pl”. NR performed the R-based graphical representation. NR drafted the manuscript and integrated substantial revisions from ADP, IM, PC, AL and CC. All authors commented and approved the final manuscript including the author’s contribution to the study, and have agreed both to be personally accountable for the author’s contributions and to ensure that questions related to the accuracy or integrity of any part of the work are appropriately investigated, resolved, and documented.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The outcomes of the present study derive from the official control activities of the Public Health Local Authority of Abruzzo region. All human data and samples were collected ethically and the need for informed consents to participates was deemed unnecessary according to national regulations (“Decreto della Giunta Regionale DGR n. 194 del 2.04.2021” from the “Dipartimento Sanità della REGIONE ABRUZZO”) and was waived by an Institutional Review Board (IRB), so-called the IRB of the National Reference Centre for Whole Genome Sequencing of microbial pathogens: database and bioinformatic analysis, because the related data and samples were openly available to the public in GISAID before the initiation of the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Metadata and PANGO lineages of the dataset of SARS-CoV-2 samples isolated by IZSAM in provinces of the Abruzzo region (Italy), then shotgun sequenced and analyzed in GENPAT until April 2021 (n = 1592).
Additional file 2.
Metadata and PANGO lineages of the dataset of SARS-CoV-2 samples isolated from several Italian provinces and retrieved by GENPAT from GISAID until April 2021 (n = 17,201).
Additional file 3.
Distributions per Italian provinces (n = 25) of the SARS-CoV-2 PANGO lineages in Italy (n = 176) retrieved by GENPAT from GISAID until April 2021 (n = 17,201).
Additional file 4.
Newick file inferred through variant calling-based analysis, binary matrix of functional annotations of variants (pan-SNPs, -MNPs and -InDels) with the program “vcf2mst.pl” and Hamming-like distance-based minimum spanning tree (MST) implemented in GrapeTree (“MSTree V2”), from the dataset of SARS-CoV-2 samples isolated by IZSAM in provinces of the Abruzzo region (Italy), then shotgun sequenced and analyzed in GENPAT until April 2021 (n = 1553).
Additional file 5.
Newick file inferred through variant calling-based analysis, binary matrix of functional annotations of variants (pan-SNPs, -MNPs and -InDels) with the program “vcf2mst.pl” and Hamming-like distance-based minimum spanning tree (MST) implemented in GrapeTree (“MSTree V2”), from the dataset of SARS-CoV-2 samples isolated from several Italian provinces and retrieved by GENPAT from GISAID until April 2021 (n = 15,451).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Di Pasquale, A., Radomski, N., Mangone, I. et al. SARS-CoV-2 surveillance in Italy through phylogenomic inferences based on Hamming distances derived from pan-SNPs, -MNPs and -InDels. BMC Genomics 22, 782 (2021). https://doi.org/10.1186/s12864-021-08112-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-08112-0