- Research
- Open access
- Published:
Word-based characterization of promoters involved in human DNA repair pathways
BMC Genomics volume 10, Article number: S18 (2009)
Abstract
Background
DNA repair genes provide an important contribution towards the surveillance and repair of DNA damage. These genes produce a large network of interacting proteins whose mRNA expression is likely to be regulated by similar regulatory factors. Full characterization of promoters of DNA repair genes and the similarities among them will more fully elucidate the regulatory networks that activate or inhibit their expression. To address this goal, the authors introduce a technique to find regulatory genomic signatures, which represents a specific application of the genomic signature methodology to classify DNA sequences as putative functional elements within a single organism.
Results
The effectiveness of the regulatory genomic signatures is demonstrated via analysis of promoter sequences for genes in DNA repair pathways of humans. The promoters are divided into two classes, the bidirectional promoters and the unidirectional promoters, and distinct genomic signatures are calculated for each class. The genomic signatures include statistically overrepresented words, word clusters, and co-occurring words. The robustness of this method is confirmed by the ability to identify sequences that exist as motifs in TRANSFAC and JASPAR databases, and in overlap with verified binding sites in this set of promoter regions.
Conclusion
The word-based signatures are shown to be effective by finding occurrences of known regulatory sites. Moreover, the signatures of the bidirectional and unidirectional promoters of human DNA repair pathways are clearly distinct, exhibiting virtually no overlap. In addition to providing an effective characterization method for related DNA sequences, the signatures elucidate putative regulatory aspects of DNA repair pathways, which are notably under-characterized.
Background
Genomic signature techniques were originally developed for identifying organism-specific characterizations [1, 2]. Genomic signature methods carry the limitation that they were not designed for sub-categorization of sequences from within a single organism. To address this shortcoming, the authors present genomic signature techniques that can be used to identify regulatory signatures, i.e. to classify DNA sequences regarding related biological units within an organism, such as particular functions, pathways and tissues.
The term genomic signature was introduced by Karlin and Burge to refer to a function characterizing genomes based on compositional variation [2]. Karlin and others showed that a di-nucleotide odds-ratio was an effective genomic signature. In addition to the odds ratio, oligonucleotide frequencies (as n-mers) and machine learning methods have been employed to classify sequences based on their organism of origin [1, 3–20], and to identify unique features of genomic data sets. Such approaches were effectively employed in a more refined focus examining tissue-specific categorization of regulatory sequences in liver or muscle [21–24].
Here, the authors employ a word-based genomic signature method. That is, given a group of related sequences, a set of characteristic subsequences is discovered. Each subsequence is called a genomic word. The set of characteristic subsequences and their attributes constitute a word-based genomic signature. It is hypothesized that each functionally related group of sequences has a detectable word-based signature, consisting of multiple genomic words. Furthermore, it is hypothesized that the genomic words that constitute a word-based genomic signature are functional genomic elements. Unlike most existing types of genomic signatures, a word-based genomic signature provides insights that are directly applicable to the problem of identifying functional DNA elements, because the words identify putative transcription factor binding sites.
The authors have identified two primary components of word-based genomic signatures that are useful for characterizing a set of related genomic sequences, RGS. The set of statistically overrepresented words that can be derived from RGS can be regarded as a word-based signature (SIG1) since it provides information about the complete set of potential control elements regulating the set of RGS. A second signature (SIG2) provides a set of words related to the elements of SIG1. The similarity between the sets can be measured based on evolutionary distance metrics, e.g. hamming and edit distance (also called Levenshtein distance, see Methods). In addition to SIG1 and SIG2 several post-processing steps built upon the two word-based signatures are undertaken to create the final regulatory genomic signature. These post-processing steps include sequence clustering, co-occurrence analysis, biological significance analysis, and a conservation analysis.
DNA repair genes represent a large network of genes that respond to DNA damage within a cell. Discrete pathways for DNA repair responses have been identified in the Reactome database [25]. A discernable feature among genes in these pathways is the promoter architecture. A large percentage of genes with DNA repair functions are regulated by bidirectional promoters [26, 27], whereas the rest are regulated by unidirectional promoters. Bidirectional promoters fall between the DNA repair gene and a partner gene that is transcribed in the opposite direction. The close proximity of the 5' ends of this pair of genes facilitates the initiation of transcription of both genes, creating two transcription forks that advance in opposite directions. DNA repair genes rarely share bidirectional promoters with other DNA repair genes. Rather, they are paired with genes of diverse functions [26].
The formal definition of a bidirectional promoter requires that the initiation sites of the genes are spaced no more than 1000 bp from one another. Using these criteria the authors have comprehensively annotated the human and mouse genomes for the presence of bidirectional promoters, using in silico approaches [26, 28]. Bidirectional promoters utilized repeatedly in the genome are known to regulate genes of a specific function [26] and serve as prototypes for complete promoter sequences for computational studies- i.e., one can deduce the full intergenic region because exons flank each side. These promoters represent a class of regulatory elements with a common architecture, suggesting a common regulatory mechanism could be employed among them. Recent molecular studies confirm that RNA PolII can dock at promoters while simultaneously facing both directions [29], rather than being restricted to a single direction.
DNA repair genes are likely to play a universal role in damage repair, therefore mutations that affect their regulation will become important diagnostic indicators in disease discovery. The authors have previously shown that bidirectional promoters regulate genes with characterized roles in both DNA repair and ovarian cancer [28]. A more detailed analysis of the regulatory motifs within this subset of promoters will address regulatory mechanisms controlling transcription of this important set of genes. This paper presents word-based genomic regulatory signatures based on statistically overrepresented oligonucleotides (6-8 mers) found in unidirectional and bidirectional promoters of genes in DNA repair pathways. The results demonstrate the effectiveness of using signatures for classifying biologically related DNA sequences. The oligonucleotides that comprise the signatures match known binding motifs from TRANSFAC [30] or JASPAR [31] databases. Furthermore, some examples overlap and agree with experimentally validated regulatory functions.
Results
The effectiveness of genomic regulatory signatures that are based on SIG1 and SIG2 was addressed by analyzing promoter sequences for genes in DNA repair pathways of humans. The promoters were divided into two classes, the bidirectional promoters and the unidirectional promoters, and distinct genomic signatures were calculated for each class. The human DNA repair pathways included 32 bidirectional promoters and 42 unidirectional promoters. Bidirectional promoters had a GC content ranging between 47.55% and 77.09% with an average of 59.87% while unidirectional promoters varied from 38.00% to 68.09%, averaging 50.84%.
Statistically overrepresented words
For each set of promoters, the statistically overrepresented words were identified. The top 25 overrepresented 8-mer words for each dataset are presented in Tables 1a and 1b, respectively (See Additional file 1 and Additional file 2 for the complete lists of words discovered in the bidirectional and unidirectional promoter set respectively). Each word is presented as an observed number or a statistical expectation, respectively, including the number of sequences the word is contained in (S or E
S
), the number of overall occurrences of the word (0 or E
S
), and a score measuring overrepresentation for the word  . Additional information such as reverse complement words, their relative positions in the list of top words, palindromic words, and p-values assessing the statistical relevance of the appearance of the word are also presented. A comparison of Tables 1a and 1b reveals that the characteristic words for the two sets are distinct, with no overlaps. The significance of the selected 25 words can be seen by comparing their scores and p-values to the scores and p-values for all words, which are plotted in Figures 1 and 2).
. Additional information such as reverse complement words, their relative positions in the list of top words, palindromic words, and p-values assessing the statistical relevance of the appearance of the word are also presented. A comparison of Tables 1a and 1b reveals that the characteristic words for the two sets are distinct, with no overlaps. The significance of the selected 25 words can be seen by comparing their scores and p-values to the scores and p-values for all words, which are plotted in Figures 1 and 2).

Score-based scatterplots. Shown here are the scatterplots for the scores of all words contained in the bidirectional promoter dataset (a) and the unidirectional promoter dataset (b) of the DNA repair pathways.

P-Value-based scatterplots. Scatterplots of the p-values for all words contained in the promoters of the DNA repair pathways exhibiting bi-directionality (a) and uni-directionality (b).
Missing words
The dataset of bidirectional promoters and unidirectional promoters contained 21,076 and 22,101 unique words of length 8, respectively, out of 65,536 unique possibilities. Thus, in each set, more than 43,000 possible words did not occur (See Additional file 3 and Additional file 4 for the complete lists of non-occurring words). The missing words in each set were enumerated, and ranked in descending order by their ES values. The top 25 missing words are shown in Tables 2a and 2b. The scatterplot of the ES values for all missing words is shown in Figure 3; note the outlier values, which correspond with the words in Tables 2a and 2b. The utility of using missing words as regulatory signatures, as reported in the literature [32, 33], was consistent with the observation of no overlapping words between bidirectional and unidirectional promoter sets.

Scatterplot of words not detected in the promoters. Scatterplots for the expected number of sequence occurrences for every word not detected in the bidirectional (a) or unidirectional (b) promoters.
Word-based clusters
For the top 2 overrepresented words, clusters were created using two different distance metrics, hamming distance and edit distance (Tables 3, 4, 5, 6, See Additional File 5, 6, 7, 8 for the complete lists of hamming distance and edit distance based clusters for bidirectional and unidirectional promoters). Each table contains the set of words that clustered around a given 'seed' word. A comparison of the sequence logos for the hamming-distance-based clusters, presented in (Figures 4, 5), shows no overlap between the two promoter sets. Similarly, no overlap existed for clusters based on edit-distance (Figures 6, 7).

Sequence logos for bidirectional promoters. Sequence logos corresponding to the word-based clusters of the top 2 overrepresented words of the bidirectional promoters. Rank 1 (a) is corresponding to the word TCGCGCCA, while Rank 2 (b) refers to TCCCGGGA.

Sequence logo for unidirectional promoters. Sequence logos corresponding to the word-based clusters of the top 2 overrepresented words of the unidirectional promoters. Rank 1 (a) is corresponding to the word ACCCGCCT, while Rank 2 (b) refers to CTTCTTTC.

Edit distance cluster for bidirectional promoters. Sequence alignments corresponding to the word-based clusters of the top 2 overrepresented words of the bidirectional promoters. For each cluster, five words were chosen based on their overall overrepresentation in the promoter set. Rank 1 (a) is corresponding to the word TCGCGCCA, while Rank 2 (b) refers to TCCCGGGA.

Edit distance cluster for unidirectional promoters. Sequence logos corresponding to the word-based clusters of the top 2 overrepresented words of the unidirectional promoters. Rank 1 (a) is corresponding to the word ACCCGCCT, while Rank 2 (b) refers to CTTCTTTC.
Sequence-based clusters
Sequences can be clustered and categorized into different families (and subfamilies). The sequence-based clusters presented here are restricted to two promoters per cluster. Sequence clustering is a measure of the co-existence of statistically overrepresented words shared between pairs of promoters as shown in Tables 7a,b. Each cluster contains IDs for the sequences that make up the cluster and the number of overrepresented words not shared within the cluster (distance). Sequences in each set were grouped into clusters based on the set of statistically overrepresented words. The shared words for the top-scoring sequence cluster of each data set were illustrated using the GBrowse environment [34] (Figures 8, 9). The visualization shows a strong positional correlation between the sequences of the top sequence cluster for the bidirectional promoters (Word: GCCCAGCC) and minor correlation between the sequences for the unidirectional promoters (Words: AGCAGGGC, GCAGGGCG).

GBrowse visualization for primary bidirectional sequence cluster. The GBrowse visualization of the two sequences for the top sequence-based cluster in the bidirectional promoter set. Shown are the words from the set of top 60 words that are detected in these two sequences.

GBrowse visualization for primary unidirectional sequence cluster. The GBrowse visualization of the two sequences for the top sequence-based cluster in the unidirectional promoter set. Shown are the words from the set of top 60 words that are detected in these two sequences.
Word co-occurrence
The promoter sets were characterized further by word co-occurrence analysis, in which word-pairs that appeared together more frequently than expected were identified. Interesting pairs of words were selected from the overrepresented words of Table 1 (Table 8a,b). Each word pair was characterized as the number of observed or expected occurrences for the word combination (S or E
S
) and a statistical overrepresentation score . No overlap was found between the bidirectional and the unidirectional set, nevertheless, the word pairs for the bidirectional promoter set achieved a higher number of sequence hits for the pairs.
Comparison of word-based properties
The distances between the scores for different word sets (Figure 10) provided a basis for discriminating among bidirectional promoters and unidirectional promoters, (Table 9 and Figure 11), whereas similarities were identified from correlated words (Table 10 and Figure 12). These tables and figures show that word-based genomic regulatory signatures can be used to describe promoter sets based on their uniqueness.

Comparison analysis: plot for complete set of words. Comparison of the words detected for the two promoter sets based on their computed overrepresentation scores.

Comparison analysis: plot for distinctive words. The words descriptive of the unidirectional promoter set (red) and the bidirectional promoter set (green). Words that are not sufficiently descriptive of either data set are eliminated from the plot.

Comparison analysis: plot for general words. The words that are significantly correlated in both data sets.
Regulatory Database Lookup
We developed a method [35] to determine if these signatures matched any known motifs from TRANSFAC or JASPAR (Table 11). The words from bidirectional promoters matched known motifs in 8/10 cases, with the words from unidirectional promoters matching known motifs in 8/10 cases as well. Compared to the consensus sequences of the known motifs, the matches were off by no more than one letter. Some of the matches corresponded to nucleotide profiles determined from collections of phylogenetically conserved, cis-acting regulatory elements [36]. Imperfect matches resulted from bases that flanked the core motifs (Table 11a, b) (see also [37]). Such events decreased the detection score to slightly above the threshold of 85% similarity. Overall, the findings in Table 11 validate that the signatures have biological relevance and suggest that the remaining signatures, which do not match known motifs could represent novel binding sites.
Conservation analysis
To address selective constraint in the word sets, sequence conservation was examined for pairs of co-occurring words. The top ten word-pairs from the unidirectional and bidirectional datasets were examined in 28-way sequence alignments using the PhastCons [38] dataset in the UCSC Human Genome Browser [39]. The results are presented in Table 12. The bidirectional promoters revealed 9/10 word sets had a record of sequence conservation in one or both words (Table 12a). The analysis of the unidirectional promoters, presented in Table 12b, showed partial conservation in only one of the word-pairs.
Biological implications
The words in the list of bidirectional promoters were examined for known biological evidence. For instance, the gene POLH has a known binding motif, TCCCGGGA, annotated as a PAX-6 binding site in the cis-RED database http://www.cisred.org/. This is the same sequence as the second most common word in the bidirectional promoters. Along with sequences that cluster with this word, we found that 19/32 genes in the bidirectional promoter set had a match to this word cluster (cluster 2) within 1 kb of their TSS, while 15/32 bidirectional promoters had a match to the words of cluster 1. Furthermore, this word also represents a Stat5A recognition site (Table 11). The RAD51 gene, which is known to be regulated by STAT5A, showed two examples from this word cluster (TGCCGGGA and TCCCGGGC).
Limitations of the approach
The presented approach does not attempt to automate the process of finding a small set of regulatory elements for a limited set of related genomic signatures like MEME [40] or AlignACE [41]. The different approach presented here produces more detailed information outside of the limited list by showing a larger (complete) set of words that are ranked based on their statistical significance. Additionally, word- and sequence-based clusters, word co-occurrences and functional significance of the words have been computed as a means of adding more detail to the retrieval of putative elements allowing a more informed interpretation of the actual regulatory function of a word.
Conclusion
This paper presents a word-based genomic signature that characterizes a set of sequences with (1) statistically overrepresented words, (2) missing words, (3) word-based clusters, (4) sequence-based clusters and (5) co-occurring words. The word-based signatures of bidirectional and unidirectional promoters of human DNA repair pathways showed virtually no overlap, thereby demonstrating the signature's utility.
In addition to providing an effective characterization method for related DNA sequences, the signatures elucidate putative regulatory aspects of DNA repair pathways. Genes in DNA repair pathways contribute to diverse functions such as sensing DNA damage and transducing the signal, participating in DNA repair pathways, cell cycle signalling, and purine and pyrimidine metabolism. The synchronization of these functions implies co-regulatory relationships of the promoters of these genes to ensure the adequate production of all the necessary components in the pathway. We present a subtle, yet detectable signature for bidirectional promoters of DNA repair genes. The consensus patterns, detected as words and related clusters of words, provide a DNA pattern that is strongly represented in these promoters. Although the proteins that bind these sequences must be examined experimentally, the data show that a protein such as STAT5A could be involved in regulating many of these promoters. STAT5A has biological relevance in DNA repair pathways, playing a known role in the regulation of the RAD51 gene. We propose that this initial study of a network of DNA repair genes serve as a model for studies that examine regulatory networks. As the relationships among genes involved in DNA repair pathways are elucidated more thoroughly, the analyses of their regulatory relationships will gain more power to detect a larger number of DNA words that are shared in common among the network of genes. The results of this analysis are supported by evidence of sequence conservation and overlap between predicted sites and known functional elements.
Methods
Two fundamental elements of word-based genomic signatures are created with the approach presented in [42, 43]. SIG1 identifies the set of statistically overrepresented words, while SIG2 represents a set of words from SIG1 that is in itself similar to the elements of SIG1, based on a specific distance measure.
The set SIG1 is computed as described in [42, 43], which is summarized as follows:
-
1.
Identify maximally repeated words of length [m, n].
-
2.
Remove low complexity words, redundant words, and words that are contained in repeat elements.
-
3.
For each word compute a 'score' that characterizes the statistical overrepresentation of the word.
-
4.
Select the words with the highest scores.
The set SIG2 is found by taking each of the elements of SIG1 and performing 'word clustering'. For each word w ∈ SIG1, this involves a two-step process:
-
1.
Construct a set (cluster) of words from RGS that have a 'distance' of no more than h from word w. Hamming distance and edit distance are used for this step.
-
2.
Construct a motif that characterizes the set of words found in step 1.
Word-based signature (SIG1)
As the foundation of the signature generation it is necessary to compute the set of distinct words Wwcin a set of input sequences S. In order to determine the statistical significance of w ∈ Wwcit is necessary to count the total number of occurrences of a given word w
j
, o
j
, as well as the number of sequences containing the word, s
j
. The occurrence information is modelled as a set of tuples  . Assuming a binomial model for the distribution of words across the input sequences, it is possible to model the total occurrence of a word w by introducing the random variable
. Assuming a binomial model for the distribution of words across the input sequences, it is possible to model the total occurrence of a word w by introducing the random variable  , where l is the complete sequence length, v the length of w, and Y
i
a binary random variable indicating if a word occurs at position i, or not, leading to the series of yes/no Bernoulli experiments. An expected value for the specific number of occurrences for a word w can then be computed as
, where l is the complete sequence length, v the length of w, and Y
i
a binary random variable indicating if a word occurs at position i, or not, leading to the series of yes/no Bernoulli experiments. An expected value for the specific number of occurrences for a word w can then be computed as  where p
w
is the probability of word w. Following a similar modelling approach, the expected number of sequences a word occurs in is given by
where p
w
is the probability of word w. Following a similar modelling approach, the expected number of sequences a word occurs in is given by  . The actual probabilities are determined by a homogenous Markov chain model of a specific order m. Based on the expected values we compute multiple scores for each word:
. The actual probabilities are determined by a homogenous Markov chain model of a specific order m. Based on the expected values we compute multiple scores for each word:
-
 : This scoring function, called SlnSES, enables the inclusion of sequence coverage into the score. A highly scored word occurs in a large percentage of sequences in the data set. It does not necessarily have to be highly significant if the overall number of occurrences is taken into account, but it is of particular use for the discovery of shared regulatory elements across multiple sequences.
: This scoring function, called SlnSES, enables the inclusion of sequence coverage into the score. A highly scored word occurs in a large percentage of sequences in the data set. It does not necessarily have to be highly significant if the overall number of occurrences is taken into account, but it is of particular use for the discovery of shared regulatory elements across multiple sequences. -
p-Value: The p-value is defined as the probability of obtaining at least as many words as the actual observed number of words:
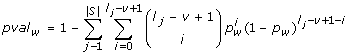 , where |S| represents the number of sequences in S and l
j
is the length of sequence j.
, where |S| represents the number of sequences in S and l
j
is the length of sequence j.
 : This scoring function, called SlnSES, enables the inclusion of sequence coverage into the score. A highly scored word occurs in a large percentage of sequences in the data set. It does not necessarily have to be highly significant if the overall number of occurrences is taken into account, but it is of particular use for the discovery of shared regulatory elements across multiple sequences.
: This scoring function, called SlnSES, enables the inclusion of sequence coverage into the score. A highly scored word occurs in a large percentage of sequences in the data set. It does not necessarily have to be highly significant if the overall number of occurrences is taken into account, but it is of particular use for the discovery of shared regulatory elements across multiple sequences.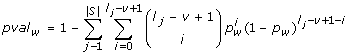 , where |S| represents the number of sequences in S and l
j
is the length of sequence j.
, where |S| represents the number of sequences in S and l
j
is the length of sequence j.Word-based clusters (SIG2)
Two methods are employed for the detection of similarities between the words that make up SIG1: hamming distance and Levenshtein distance (also called edit distance). While hamming distance is defined as the number of positions for which the corresponding characters of two words of the same length differ, edit distance allows the comparison of different length words and accounts for three edit operations (insert, delete and substitute), rather than the plain mismatch (corresponds to substitute) employed by the hamming distance.
The biological reasoning for employing distance metrics in order to group similar words together can be found in the evolution of sequences. A biological structure is constantly exposed to mutation pressure. These mutations can occur as insertions, deletions or substitutions, however insertions and deletions are deleterious in most cases, leading to the issue that edit distance provides a very detailed model of the mutations but hamming distance is a reasonable abstraction and will work well for this case. The motif logos for the hamming distance clusters were constructed using the TFBS Perl module by Lenhard and Wasserman [44]. ClustalW2 [45] was used to align the words of the edit distance clusters.
Sequence clustering
The sequence clustering conducted in this research is focussed on the words shared between element of a set of sequences. A set of words is taken as the input for the clustering. A binary vector s i = (si,1, si,2,..., si, k) for each sequence s i is created, marking an element si, kwhere k is the number of words used to distinguish the sequences with k ≤ |Wwc|. The element si, kof the vector is populated with a '1' if the word k is found in sequence i, and '0' if it is not. The similarity between sequences is determined by the dot product between the binary sequence vectors, and is deducted from the complete number of words in the vector space. In order to determine the distance between k sequences (with k ≥ 2), the dot product is extended to accommodate multiple sequences.

The cluster with the smallest distance is visualized using GMOD's GBrowse framework [34]. For each of the sequences contained in the cluster, the words pertaining to SIG1 are displayed.
Biological significance (lookup)
Once genomic signatures are identified, the next step is to discern their biological role. One important aspect of this role, crucial to understanding gene regulation [46], is the location of the preferred binding sites for certain proteins (transcription factor binding sites or TFBSs). To locate these sites, the signatures are compared to a set of known binding sites, which are usually represented as weighted matrices [47]. However, a simple scoring scheme can misclassify results when applied to the typically short sequences produced by signature finders. In this simple approach, short signatures are aligned to each matrix by ignoring the parts of the matrices that are longer than the signature. This results in erroneous scores since a signature could match just the very end of large matrix, which is often of little significance (the core of the matrix generally represents the sites of strongest binding).
To give a more significant measure of similarity, we developed a tool that uses a window around the original sequences (those which the signature is based upon) to improve the comparison. The naive implementation of this approach is to use a window of base pairs around each signature and find the optimal alignment to each TFBS matrix by scoring every possible sub-sequence containing the signature. For instance, if a signature is located 10 times within the set of sequences, each matrix is aligned to each of the 10 loci containing the signatures. Our tool uses a faster approach; it finds all occurrences of TFBSs meeting the desired threshold in every sequence, and subsequently uses this information to quickly score the signatures. As a benefit, the list of TFBS can be reused to quickly score new signatures or to redo the analysis with interesting subsets of sequences, such as all sequences which in liver cells are highly expressed.
Co-occurrence analysis
The co-occurrence analysis aims to determine the expected number of sequences containing a given pair of not necessarily distinct words at least once. If n denotes the word length, m the number of sequences,  the probability for a word i to occur anywhere in the sequence, and l
k
the length of sequence k, the expected number of sequences containing a given pair of words can be calculated as:
the probability for a word i to occur anywhere in the sequence, and l
k
the length of sequence k, the expected number of sequences containing a given pair of words can be calculated as:

The  score is used as the main scoring function in the co-occurrence analysis.
score is used as the main scoring function in the co-occurrence analysis.
Conservation analysis
Sequence conservation was mapped using PhastCons conservation scores [38] calculated on 28 species, which are based on a two-state (conserved state vs. Non-conserved region) phylo-HMM. PhastCons scores were obtained from the UCSC Human Genome Browser [39]. The scores reported by the UCSC Human Genome Browser contain transformed log-odds scores, ranging from 0–1000. Conserved regions were required to cover the majority of the word length.
Comparison
Words can have significantly different scores for each of the data sets in which they occur. In order to analyze the words based on their impact on the data sets it is useful to assign a distance metric that determines which data set is described best by a given word. Based on a graphical analysis, three points of interest can be determined: the point where the perpendicular of a given point on the x-axis crosses the main diagonal, the point where the perpendicular of a given point on the main diagonal crosses the main diagonal and finally the point where the perpendicular from a given point on the y-axis crosses the main diagonal. Based on the conventional techniques of fold-change detection in microarray analysis, we consider the perpendicular on the main diagonal. The resulting distance formula is:  , with y0 being the score for the word within the unidirectional data set, and x0 being the score of the word in the bidirectional data set.
, with y0 being the score for the word within the unidirectional data set, and x0 being the score of the word in the bidirectional data set.
References
Deschavanne PJ, Giron A, Vilain J, Fagot G, Fertil B: Genomic signature: characterization and classification of species assessed by chaos game representation of sequences. Mol Biol Evol. 1999, 16 (10): 1391-1399.
Karlin S, Burge C: Dinucleotide relative abundance extremes: a genomic signature. Trends Genet. 1995, 11 (7): 283-290.
Abe T, Kanaya S, Kinouchi M, Ichiba Y, Kozuki T, Ikemura T: A novel bioinformatic strategy for unveiling hidden genome signatures of eukaryotes: self-organizing map of oligonucleotide frequency. Genome Inform. 2002, 13: 12-20.
Abe T, Kanaya S, Kinouchi M, Ichiba Y, Kozuki T, Ikemura T: Informatics for unveiling hidden genome signatures. Genome Res. 2003, 13 (4): 693-702.
Bastien O, Lespinats S, Roy S, Metayer K, Fertil B, Codani J, Marechal E: Analysis of the compositional biases in Plasmodium falciparum genome and proteome using Arabidopsis thaliana as a reference. Gene. 2004, 336 (2): 163-173.
Bentley SD, Parkhill J: Comparative genomic structure of prokaryotes. Annu Rev Genet. 2004, 38: 771-792.
Campbell AM, Mrazek J, Karlin S: Genome signature comparisons among prokaryote, plasmid, and mitochondrial DNA. Proc Natl Acad Sci. 1999, 96 (16): 9184-9189.
Carbone A, Kepes F, Zinovyev A: Codon bias signatures, organization of microorganisms in codon space, and lifestyle. Mol Biol Evol. 2005, 22 (3): 547-561.
Deschavanne PJ, Giron A, Vilain J, Dufraigne C, Fertil B: Genomic signature is preserved in short DNA fragments. IEEE International Symposium on Bioinformatics and Biomedical Engineering. 2000
Elhai J: Determination of bias in the relative abundance of oligonucleotides in DNA sequences. J Comput Biol. 2001, 8 (2): 151-175.
Fertil B, Massin M, Lespinats S, Devic C, Dumee P, Giron A: GENSTYLE: exploration and analysis of DNA sequences with genomic signature. Nucleic Acids Res. 2005, 33: 512-515.
Gentles AJ, Karlin S: Genome-scale compositional comparisons in eukaryotes. Genome Res. 2001, 11 (4): 540-546.
Jeffrey H: Chaos game representation of gene structure. Nucleic Acids Res. 1990, 18 (8): 2163-2170.
Karlin S: Global dinucleotide signatures and analysis of genomic heterogeneity. Curr Opin Microbiol. 1998, 1 (5): 598-610.
Karlin S, Campbell AM, Mrazek J: Comparative DNA analysis across diverse genomes. Annu Rev Genet. 1998, 32: 185-225.
Karlin S, Mrazek J, Campbell AM: Compositional biases of bacterial genomes and evolutionary implications. J Bacteriol. 1997, 179 (12): 3899-3913.
Karlin S, Mrazek J, Gentles AJ: Genome comparisons and analysis. Curr Opin Struct Biol. 2003, 13 (3): 344-352.
Li J, Sayood K: A Genome Signature Based on Markov Modeling. Proceedings of the 27th Annual International Conference of the IEEE – Engineering in Medicine and Biology Society:. 2005, 2005- ; Shanghai
Wong K, Finan TM, Golding GB: Dinucleotide compositional analysis of Sinorhizobium meliloti using the genome signature: distinguishing chromosomes and plasmids. Funct Integr Genomics. 2002, 2 (6): 274-281.
Zhang C, Zhang R, Ou H: The Z curve database: a graphic representation of genome sequences. Bioinformatics. 2003, 19 (5): 593-599.
Fickett JW, Wasserman WW: Discovery and modeling of transcriptional regulatory regions. Curr Opin Biotechnol. 2000, 11 (1): 19-24.
Schones DE, Sumazin P, Zhang MQ: Similarity of position frequency matrices for transcription factor binding sites. Bioinformatics. 2005, 21 (3): 307-313.
Wasserman WW, Fickett JW: Identification of regulatory regions which confer muscle-specific gene expression. J Mol Biol. 1998, 278 (1): 167-181.
Wasserman WW, Palumbo M, Thompson W, Fickett JW, Lawrence CE: Human-mouse genome comparisons to locate regulatory sites. Nat Genet. 2000, 26 (2): 225-228.
Joshi-Tope G, Gillespie M, Vastrik I, D'Eustachio P, Schmidt E: Reactome: a knowledgebase of biological pathways. Nucleic Acids Res. 2005, 33: D428-432.
Yang MQ, Elnitski L: A computational study of bidirectional promoters in the human genome. Springer Verlag Lecture Notes in Bioinformatics. Edited by: Zhang Y Heidelberg. 2007, 361-371.
Adachi N, Lieber MR: Bidirectional gene organization: a common architectural feature of the human genome. Cell. 2002, 109: 807-809.
Yang MQ, Koehly LM, Elnitski LL: Comprehensive annotation of bidirectional promoters identifies co-regulation among breast and ovarian cancer genes. PLoS Comput Biol. 2007, 3 (4): e72-
Seila AC, Calabrese JM, Levine SS, Yeo GW, Rahl PB, Flynn RA, Young RA, Sharp PA: Divergent Transcription from Active Promoters. Science. 2008, 1162253-
Wingender E, Chen X, Hehl R, Karas H, Liebich I: TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res. 2000, 28: 316-319.
Bryne JC, Valen E, Tang MH, Marstrand T, Winther O: JASPAR, the open access database of transcription factor-binding profiles: new content and tools in the 2008 update. Nucleic Acids Res. 2008, 36: D102-106.
Herold J, Kurtz S, Giegerich R: Efficient computation of absent words in genomic sequences. BMC Bioinformatics. 2008, 9 (1): 167-
Hampikian G, Andersen T: Absent sequences: nullomers and primes. Pac Symp Biocomput. 2007, 355-366.
Stein LD, Mungall C, Shu S, Caudy M, Mangone M, Day A, Nickerson E, Stajich JE, Harris TW, Arva A, et al: The generic genome browser: a building block for a model organism system database. Genome Res. 2002, 12 (10): 1599-1610.
Jacox E, Elnitski L: Finding Occurrences of Relevant Functional Elements in Genomic Signatures. International Journal of Computational Science. 2008
Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V: Systematic discovery of regulatory motifs in human promoters and 3' UTRs by comparison of several mammals. Nature. 2005, 434: 338-345.
Chekmenev DS, Haid C, Kel AE: P-Match: transcription factor binding site search by combining patterns and weight matrices. Nucleic Acids Res. 2005, 33: W432-437.
Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al: Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15 (8): 1034-1050.
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler aD: The Human Genome Browser at UCSC. Genome Res. 2002, 12 (6): 996-1006.
Bailey TL, Williams N, Misleh C, Li WW: MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006, 34: W369-373.
Roth FP, Hughes JD, Estep PW, Church GM: Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat Biotech. 1998, 16 (10): 939-945.
Lichtenberg J, Jacox E, Yang MQ, Elnitski L, Welch L: Word-based characterization of the bidirectional promoters from the human DNA-repair pathways. The 2008 International Conference on Bioinformatics and Computational Biology. 2008, Las Vegas
Lichtenberg J, Morris P, Ecker K, Welch L: Discovery of regulatory elements in oomycete orthologs. The 2008 International Conference on Bioinformatics and Computational Biology. 2008, Las Vegas
Lenhard B, Wasserman WW: TFBS: Computational framework for transcription factor binding site analysis. Bioinformatics. 2002, 18: 1135-1136.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al: ClustalW and ClustalX version 2.0. Bioinformatics. 2007, 23 (21): 2947-2948.
Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE: Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007, 447 (7146): 799-816.
Wasserman WW, Sandelin A: Applied bioinformatics for the identification of regulatory elements. Nat Rev Genet. 2004, 5 (4): 276-287.
Acknowledgements
The Ohio University team acknowledges the support of the Stocker Endowment, Ohio University's Graduate Research and Education Board (GERB), the Ohio Plant Biotechnology Consortium, the Ohio Supercomputer Center, and the Choose Ohio First Initiative of the University System of Ohio.
The Ohio University team further acknowledges Sarah Wyatt for providing the initial motivation and guidance for the work in regulatory genomics as well as Mo Alam, Jasmine Bascom, Kaiyu Shen, Nathaniel George, Dazhang Gu, Eric Petri and Haiquan Zhang for their support during the development of the approach.
LE is supported by the Intramural Program of the National Human Genome Research Institute.
The authors would like to thank to anonymous reviewers for their insights and comments.
This article has been published as part of BMC Genomics Volume 10 Supplement 1, 2009: The 2008 International Conference on Bioinformatics & Computational Biology (BIOCOMP'08). The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2164/10?issue=S1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
JL contributed in the development of algorithms and models, the implementation of algorithms, generation of the signature data and drafting of the document. EJ contributed the lookup of biological significance for the words of the signatures. JDW contributed in the development of the models and algorithms and the implementation of the approaches. KK contributed in the development and implementation of models and algorithms. XL contributed in the development of the models and algorithms for co-occurrence analysis and generated the respective data. MQY and LE generated and categorized the promoter data set. FD contributed in the development of models and algorithms, and in the implementation of the methods. KE contributed the idea of hamming-distance-based clustering. SSL contributed to the statistical foundations of the scoring model. LE provided the text describing the biological background and significance, conducted the conservation analysis, and participated in the drafting of the paper. In addition to architecting the software pipeline employed in this research, LRW contributed to the design, implementation and validation of models and algorithms (especially in the areas of word searching, word scoring, and sequence clustering) and to the writing of this manuscript.
Electronic supplementary material
12864_2009_2546_MOESM1_ESM.csv
Additional file 1: Words discovered in bidirectional promoters. Entire set of words discovered in the bidirectional promoters with occurrences, expected occurrences, scores, reverse complement information and p-value. (CSV 1 MB)
12864_2009_2546_MOESM2_ESM.csv
Additional file 2: Words discovered in unidirectional promoters. Entire set of words discovered in the unidirectional promoters with occurrences, expected occurrences, scores, reverse complement information and p-value. (CSV 1 MB)
12864_2009_2546_MOESM3_ESM.csv
Additional file 3: Missing words in bidirectional promoters. Set of words not detected in the bidirectional promoters with expected occurrences. (CSV 1 MB)
12864_2009_2546_MOESM4_ESM.csv
Additional file 4: Missing words in unidirectional promoters. Set of words not detected in the unidirectional promoters with expected occurrences. (CSV 1 MB)
12864_2009_2546_MOESM5_ESM.csv
Additional file 5: Hamming distance clusters in bidirectional promoters. Entire set of hamming distance based clusters for the bidirectional promoters with detailed cluster element information position weight matrix and corresponding regular expression motif. (CSV 68 KB)
12864_2009_2546_MOESM6_ESM.csv
Additional file 6: Hamming distance clusters in unidirectional promoters. Entire set of hamming distance based clusters for the unidirectional promoters with detailed cluster element information position weight matrix and corresponding regular expression motif. (CSV 51 KB)
12864_2009_2546_MOESM7_ESM.csv
Additional file 7: Edit distance clusters in bidirectional promoters. Entire set of edit distance based clusters for the bidirectional promoters with detailed cluster element information position weight matrix and corresponding regular expression motif. (CSV 751 KB)
12864_2009_2546_MOESM8_ESM.csv
Additional file 8: Edit distance clusters in unidirectional promoters. Entire set of edit distance based clusters for the unidirectional promoters with detailed cluster element information position weight matrix and corresponding regular expression motif. (CSV 620 KB)
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lichtenberg, J., Jacox, E., Welch, J.D. et al. Word-based characterization of promoters involved in human DNA repair pathways. BMC Genomics 10 (Suppl 1), S18 (2009). https://doi.org/10.1186/1471-2164-10-S1-S18
Published:
DOI: https://doi.org/10.1186/1471-2164-10-S1-S18